

Giant viruses
Giant viruses have genomes that can reach multiple megabases in length and carry complex functional traits that led to a reexamination of the place of viruses in the tree of life [1, 2, 3]. Some giant viruses build particles so big they might be confused with bacterial cells. In fact, the first description of a giant virus back in 2003 was delayed by 10 years because researchers initially assumed that what would eventually be named “Acanthamoeba polyphaga mimivirus” (for “Mimicking Microbes”) was a cryptic bacteria infecting amoeba yet resisting traditional bacterial identification protocols [4, 5, 6]. Contemplating electron microscopy results revealing the replication cycle of a virus of unprecedented size must have been an electrifying Eureka moment for La Scola and colleagues. Hundreds of publications later, the giant viruses and their numerous close relatives are commonly referred to as Nucleocytoplasmic Large DNA Viruses (NCLDVs), have formed the phylum Nucleocytoviricota within the Varidnaviria realm [7], and were found to collectively infect the entire eukaryotic domain with at times major economical consequences (e.g., hemorrhagic fever in swine). It is now well demonstrated that NCLDVs have a profound influence on the ecology (e.g., regulation of oceanic blooms) and evolution (e.g., gene flow) of various eukaryotes. In addition, some researchers actively explore how NCLDVs may have been driving the emergence of complex proto-eukaryotic traits (viral eukaryogenesis hypothesis) in a very distant past [8, 9, 10].
NCLDVs are frequently part of metagenomic assemblies and their corresponding metagenome-assembled genomes (MAGs) have been characterized, at least to some extent, using automatic binning algorithms and the occurrence of few hallmark genes for guidance [10, 11]. These prolific surveys, alongside co-culture efforts, have provided considerable insights into the gene pool, diversity and evolution of the phylum Nucleocytoviricota. Notably, this phylum was organized into two major classes, Pokkesviricetes and Megaviricetes, the later being possibly more diverse than bacterial populations in the oceans [13]. As observed for the three domains of life in the last decade, our global understanding of NCLDVs will most likely strongly benefit from the characterization of many more environmental genomes in years to come. Yet, very few researchers have characterized NCLDV MAGs thus far. Hence, the genomic characterization of giant viruses and their close relative would strongly benefit from a democratization of genome-resolved metagenomics focused on these fascinating targets. This blog post describes one way to manually identify and bin NCLDVs from metagenomic projects. It was written for those contemplating this environmental genomic journey into the unknowns, beyond the stricto sensus tree of life, in the land of viral gigantism that has puzzled scientists since the early 2000s.
Tara Oceans Data and the “classical” anvi’o binning framework
The anvi’o programs and visualization capabilities dedicated to genome-resolved metagenomics [14, 15] have empowered microbiologists and other binning enthusiasts to characterize and assess the quality of MAGs corresponding to Bacteria, Archaea and Eukarya without the need for extensive bioinformatic skills. For example, the considerable metagenomic legacy of Tara Oceans [16] was used from within anvi’o to characterize marine MAGs from the three domains of life [17, 18, 19]. Giant viruses were not considered in these surveys, in part because there was no method in place to properly identify them. To fill this gap, a simple framework focused on some relevant genes was recently developed and subsequently used to characterize nearly seven hundred NCLDV MAGs abundant in the sunlit ocean [20]. The study exposed two additional classes of NCLDVs: Proculviricetes and Mirusviricetes. Here, examples from this survey are used to describe in more details how MAGs for giant viruses and their close relatives can be easily characterized from within the anvi’o interactive interface using ready-to-use Hidden Markov Models (HMMs).
Tara Oceans metagenomes have been co-assembled one oceanic region at a time for binning purposes [18]. Metagenomic assembly subsets were generated from this data using constrained binning based on sequence composition and differential coverage (CONCOCT algorithm). Self-contained anvi’o contigs and profile databases were generated for each subset. Here we first take the example of one of these subsets (PSW_Bin_158), corresponding to one region of the Pacific Ocean and containing about 2,000 contigs. Using the “classical” anvi’o genome-resolved metagenomics workflow, bacterial MAGs displaying coherent differential coverage values across the metagenomic samples considered could be identified (Figure 1) and subsequently used to study the genomics of plankton [19]. Yet, some clusters of contigs did not appear to represent substantial portions of microbial genomes (anvi’o could not find sufficient single-copy core genes from the bacterial, archaeal and eukaryotic collections), and were left out of the survey. This is a very common limitation of genome-resolved metagenomics: not all contigs end up in a MAG. Part of the problem is that we lack proper methodologies to make sense of all biologically relevant metagenomic bins. For example, eukaryotic MAGs looked like poorly resolved archaeal genomes until a relevant collection of eukaryotic single-copy core genes was integrated into the anvi’o code base.
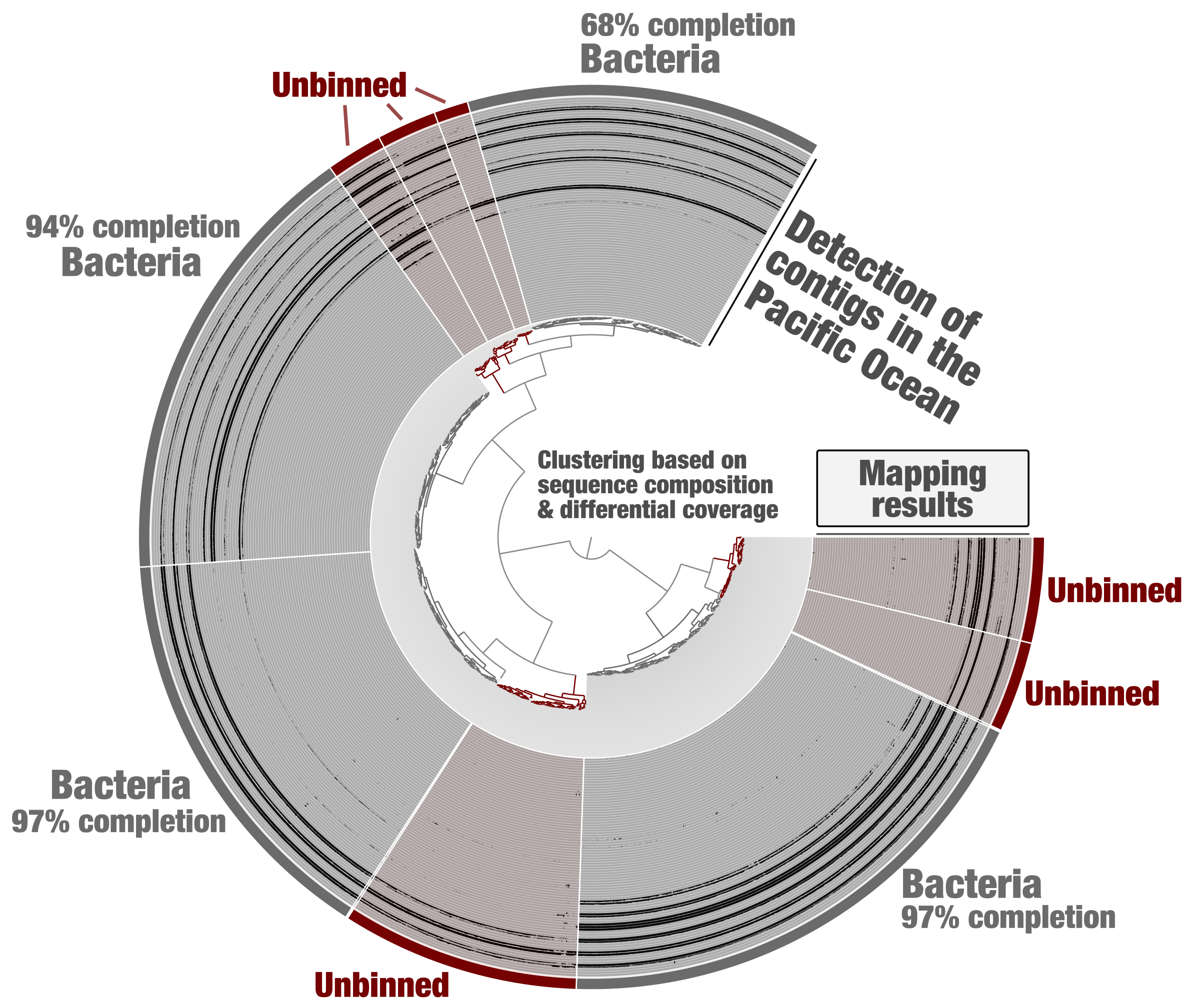
Figure 1: Identification of bacterial MAGs from within the anvi’o interactive interface. Contigs from the metagenomic subset PSW_Bin_158 were clustered based on sequence composition and differential coverage across Tara Oceans metagenomes of the Pacific South West. Detection of contigs across metagenomes is displayed as individual layers (mapping results, minimum value set to 0.25). Bins were selected manually from the interface.
Binning MAGs of giant viruses and their close relatives with anvi’o
In order to identify putative NCLDV MAGs from the metagenomic assembly subset PSW_Bin_158, a HMM screening was done to search for 8 NCLDV hallmark genes (those are present in most reference genomes, and a majority of them are usually single-copy core genes) and 149 NCLDV core genes (those occur in some of the reference genomes). These HMMs were previously built by Julien Guglielmini, Morgan Gaia, Patrick Forterre and their colleagues to study the evolutionary history of giant viruses and their close relatives [10], using in part previously identified NCLDV conserved genes dubbed ncVOGs [21]. Within anvi’o, HMMs were used for each NCLDV hallmark gene so that their signal would later be displayed as individual layers in the interactive interface (program “anvi-run-hmms” with the flag “-H”). A single HMM for the 149 NCLDV core genes was then used (same program) so that their cumulative signal would be displayed as one additional layer. Based on the NCLDV HMM hits, one could easily identify a cluster of contigs that (1) displays coherent differential coverage values across samples, (2) contains all the NCLDV hallmark genes (most being single-copy), (3) and is substantially enriched in NCLDV core genes as compare to the rest of PSW_Bin_158 (Figure 2). Subsequent phylogenetic analyses [20] indicated that this giant virus MAG (TARA_PSW_NCLDV_00001), with a length of 1.45 Mbp, corresponds to the order Imitervirales (class Megaviricetes), a well-known lineage of NCLDVs highly diverse in the sunlit ocean.
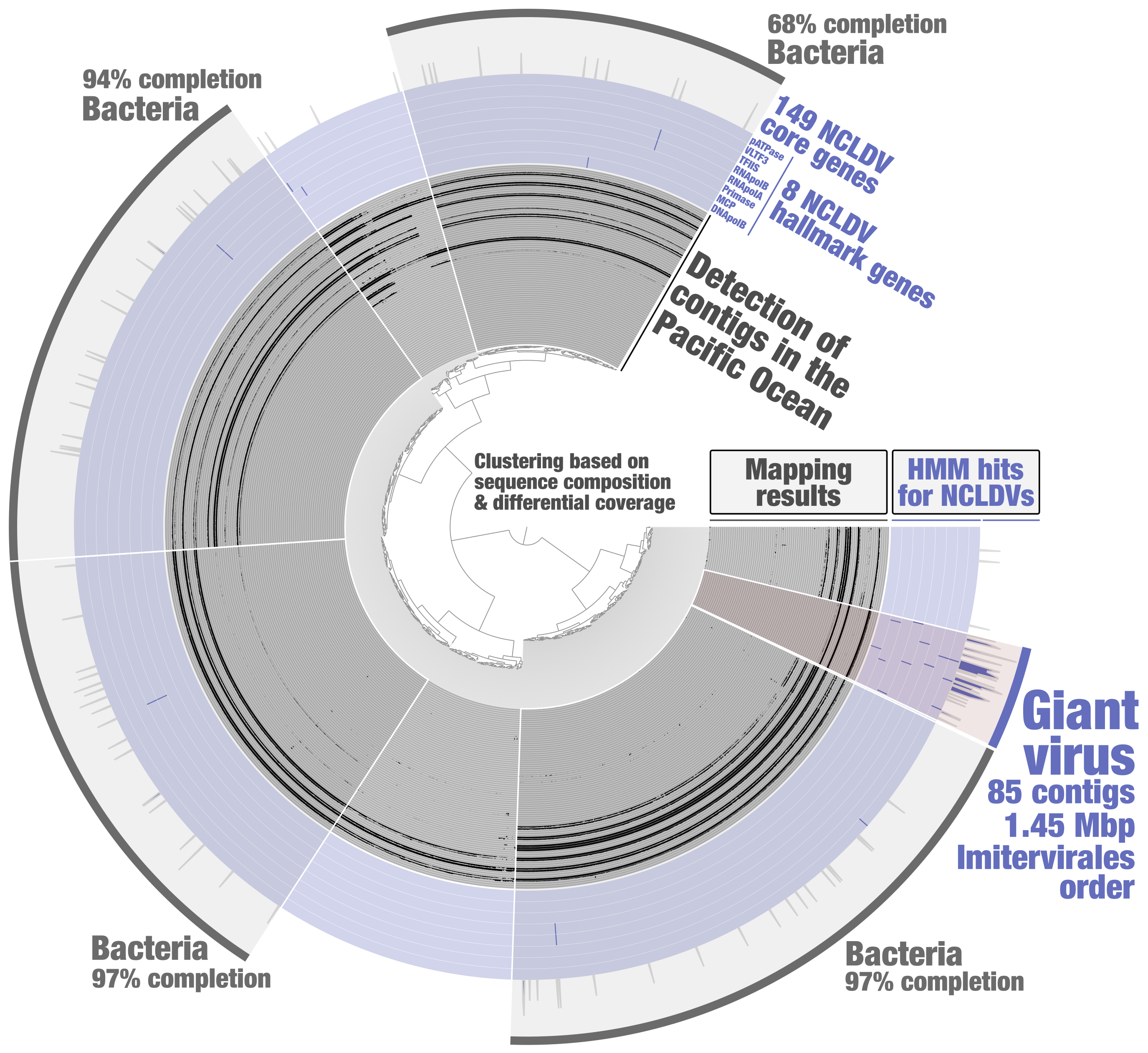
Figure 2: Identification of a giant virus MAG from within the anvi’o interactive interface. Contigs from the metagenomic subset PSW_Bin_158 were clustered based on sequence composition and differential coverage across Tara Oceans metagenomes of the Pacific South West. Detection of contigs across metagenomes is displayed as individual layers (mapping results, minimum value set to 0.25). Bins were selected manually from the interface.
While only one NCLDV MAG was present in the subset PSW_Bin_158, other subsets contained many more NCLDVs and were therefore slightly more challenging to bin. In the example of the metagenomic subset ARC_136 (Arctic Ocean) for instance, two closely related clusters of contigs displayed signal for NCLDVs. Should they be binned together or separately? Even though there is currently no comprehensive bioinformatic tool available to estimate the genomic completion and redundancy of giant viruses and their close relatives, some of the hallmark genes found in single-copy across most lineages (e.g., TFIIS) can be highly valuable in such situation. Here, the hallmark genes pointed to the occurrence of two distinct NCLDV MAGs (Figure 3). Thus, these genes could be used, at least to some extent, for binning guidance and to assess the quality of NCLDV MAGs. Their occurrence suggests that the two selected MAGs (length of 321 Kbp and 377 Kbp) are of relatively good completion and represent individual NCLDV population genomes.

Figure 3: Binning two NCLDV MAGs with similar distribution patterns. The figure is a screenshot of a section of the anvi’o interactive interface when binning the metagenomic subset ARC_Bin_136 (Arctic Ocean). Following the same display as in Figure 2, gray layers correspond to the detection of contigs across metagenomes, and the blue layers correspond to the detection of NCLDV hallmark genes (first 8 layers) and 149 NCLDV core genes (last layer). Bins were selected manually from the interface.
Perspective: assessing the completion and redundancy of giant virus MAGs
Currently, anvi’o does not provide real-time completion and redundancy estimates for the NCLDVs. One challenge is that single-copy core genes vary substantially between NCLDV lineages. For instance, the newly discovered Mirusviricetes class lacks 50% of the hallmark genes and exposed a previously overlooked functional life style for giant virus relatives. Another challenge is that we have very few complete NCLDV genomes from culture (most are MAGs of undetermined completion). Nevertheless, we are working on identifying as many lineage specific single-copy core genes as possible, in part using gene clusters of remote homologies with AGNOSTOS [22], to address the problem of completion and redundancy estimates. The anvi’o developers might then integrate this feature into the code base in future releases, provided that sufficiently good results are obtained. One likely scenario is that various single-copy core gene collections will be needed for the NCLDVs, contrasting with the three domains of life that require a single collection each. In the best-case scenario, random forest based on the occurrence of the single-copy core genes will be able to properly link MAGs to NCLDV lineages and use the corresponding collection to estimate their completion and redundancy, in real time during binning or when processing MAG collections. A possible alternative would be to identify the lineage based on BLAST results (similar to anvi-estimate-scg-taxonomy) for some of the hallmark genes prior to using the corresponding collection of single-copy core genes. These methodological advances could help democratizing the environmental genomic exploration of NCLDVs by microbiologists and other binning enthusiasts, without the need for particular bioinformatic skills.
Data availability
The anvi’o compatible user-defined hmm-source files, as well as contigs and profile databases that correspond to the metagenomic assembly subsets used as examples here are available at doi:10.6084/m9.figshare.17712038. Those files can be used to reproduce the binning results presented and to explore NCLDVs in other metagenomic data, provided anvi’o v7.1 (or a more recent version) is installed.