

Table of Contents
The study Hug et al. published last week puts the genomic discoveries Banfield group made during the last years into a striking contex with a beautiful title, “A new view of the tree of life”:
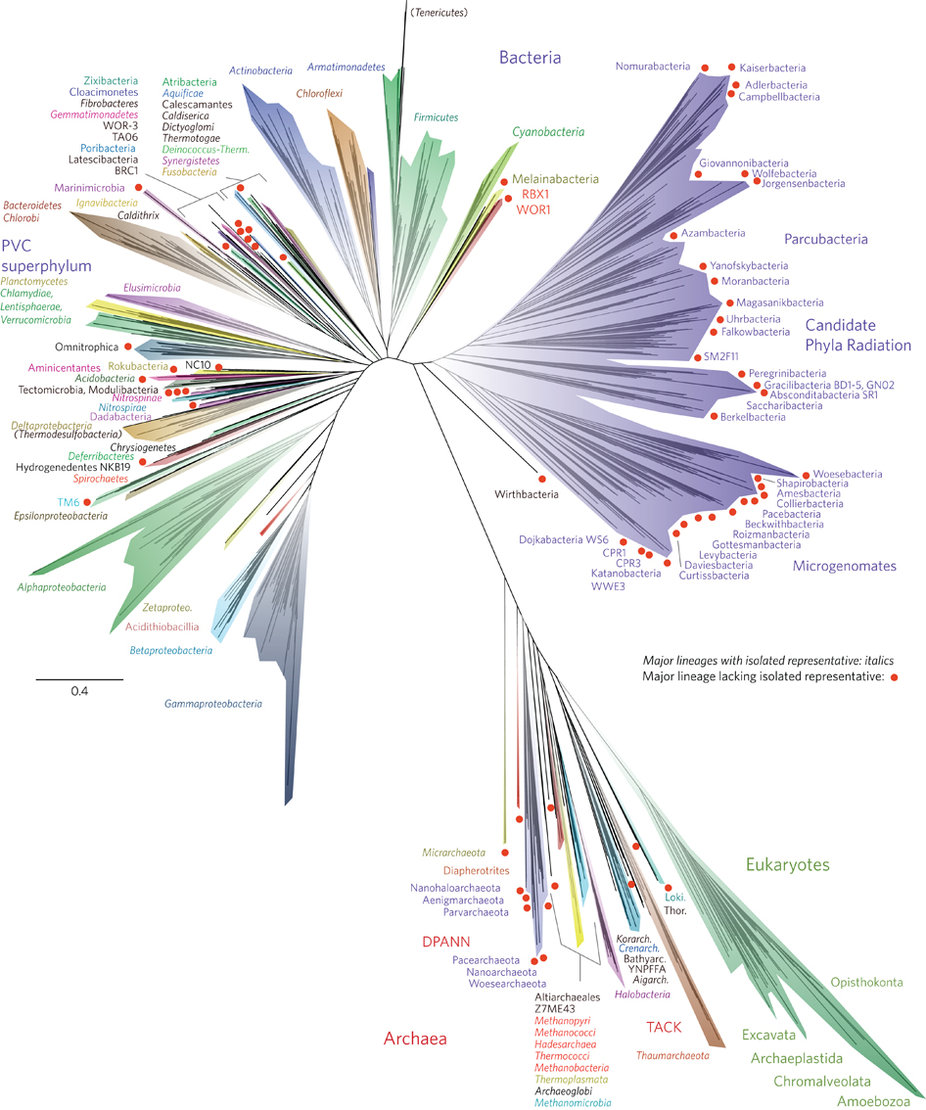
The significance of this tree has been covered broadly by many, including Ed Yong, and Carl Zimmer, so I will save you from another attempt that will definitely not match to their’s.
Briefly, the tree includes a total of 3,083 genomes, 1,011 of which are the unusual genomes authors identified from various environments using assembly-based shotgun metagenomics and binning. The phylogenetic inference in the figure is based on 16 ribosomal proteins from each organism, and so on.
A large fraction of the genomes in Hug et al.’s study comes from another publication from the Banfield group which appeared in Nature less than a year ago: Unusual biology across a group comprising more than 15% of domain Bacteria. This study describes 797 tiny genomes, most of which lack various pathways, which makes them very hard to cultivate, and contain quirky ribosomal operons, which renders 16S rRNA gene-based surveys unable to detect them. In their paper Brown et al. describes this group of more than 35 new phyla as the “candidate phyla radiation” (CPR). I will mostly focus on these genomes in this post, to make a point about the rest of CPR.
Distribution of bacterial single-copy genes in CPR
The use of bacterial single-copy genes is quite a common technique to get a ballpark estimation of how much of a genome is captured in a metagenomic bin. They are quite useful. In fact, they can even be used to estimate the number of bacterial genomes in an assembly. Instead of 51 single-copy genes the Banfield group normally uses, the 2015 Brown et al. study employs 43 bacterial single-copy genes to estimate the level of completeness of their CPR genomes since they already knew some genes were absent in CPR genomes. Here I re-analyzed these 797 genomes by extending the number of single-copy genes to 139, by using Campbell et al.’s bacterial single-copy gene collection (details of which is in the supplementary table 2, and we have them nicely organized in our codebase). To create this collection, Campbell et al. analyzed the occurrence of protein families in 1,516 complete bacterial genomes distributed around bacterial tree of life, and identified protein families that occurred only once in at least 90% of all genomes. For the bacterial life we knew about, it works beautifully well. But how well are these genes represented in Brown et al’s collection?
Here is an anvi’o figure to answer that question visually:
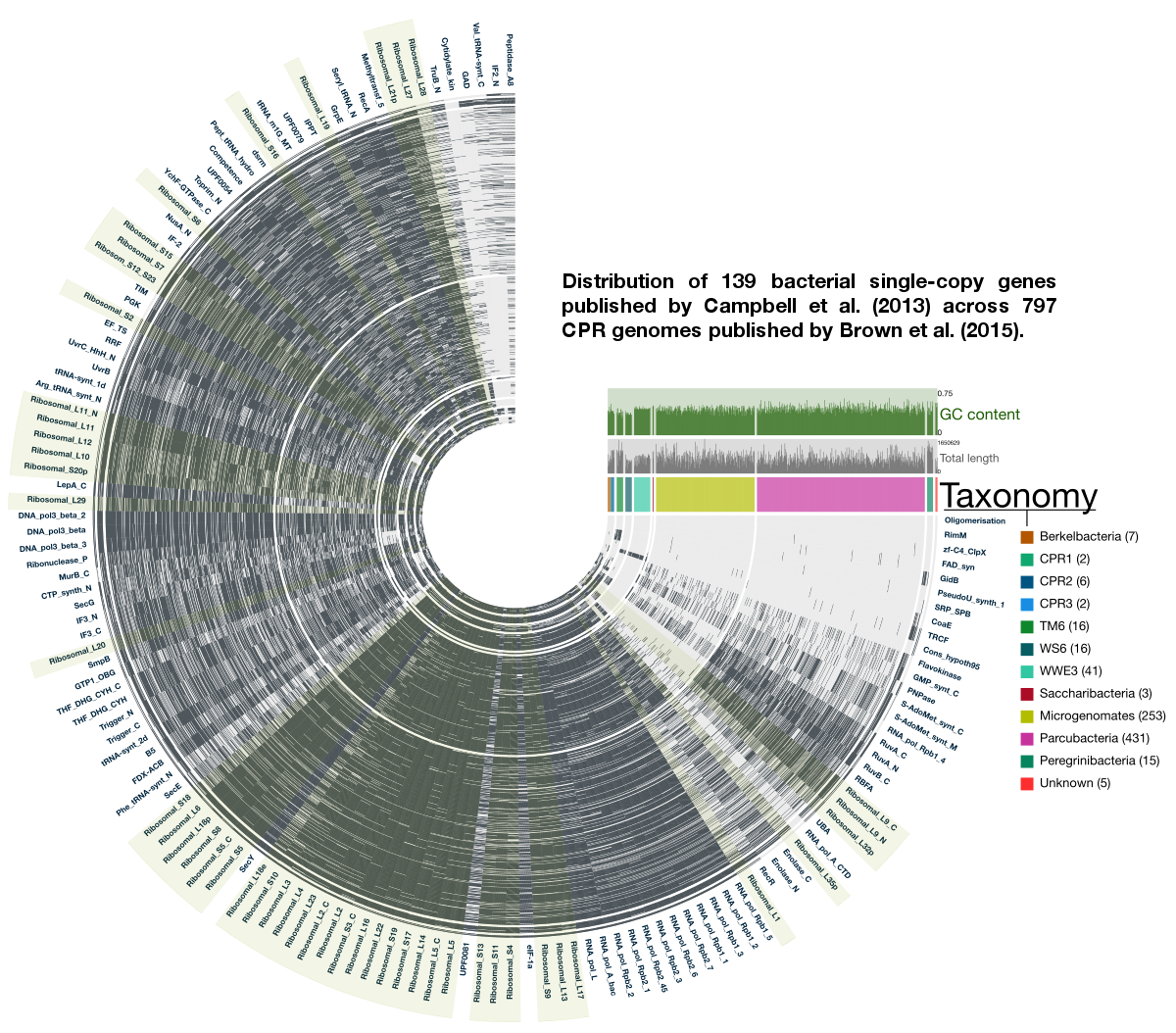
In this figure, each radial layer represents one of the 797 genomes, each tip of the tree represents a bacterial single-copy gene, and each black bar indicates whether a given single-copy gene was present (based on HMM profile matches beyond model noise cut-off identified by Campbell et al.).
The good news: This display agrees with Brown et al.’s findings. The not-yet-good-or-bad news: it also extends the list of single-copy genes CPR genomes lack.
The taxonomy Brown et al. identified has a pretty striking signal with respect to how certain groups of genomes lack certain bacterial single-copy genes completely: a small set of ribosmal proteins does contain enough signal for a phylogenetic analysis to organize genomes in a manner that is coherent with the rest of the genome. This is a simple, but very convincing boost in confidence. Because if one thinks careful enough, they may realize that it may not have been the case at all.
I find this even more striking: these single-copy genes occur in more than 90% percent of bacterial genomes, yet patterns emerge from their presence/absence across CPR genomes. The non-uniform presence/absence patterns of these core genes between groups of CPR genomes suggests an even richer level of diversity in this clade we had not seen before… What we have seen so far is probably the tip of the iceberg.
OK. Can this information be useful?
Training a classifier to predict CPR genomes
Clearly these genomes represent quite a large fraction of the diversity of life on the planet (“comprising more than 15% of domain Bacteria”, as the authors put it). Then, people who do metagenomic binning must have been recovering them, too.
That being said, their small size and low completion scores in general do not make them “interesting” targets in the conventional sense of the word, making them easy to discard as poorly identified bacterial genomes from metagenomic binning results unless the researcher has not been looking for them specifically. Nevertheless, probably they were hidden in plain sight, even for researchers who are dedicated to characterize bacterial life in their system beyond 16S surveys or annotation of short metagenomic reads.
If there was a way to recognize CPR genomes rapidly, maybe it would have been useful to understand the extent of their distribution among metagenomic bins, and help people to focus on the ones that look interesting. And this is exactly what we will explore here using those single-copy gene patterns.
Essentially we want to say “if these single-copy genes are lacking from a genome, and those single-copy genes are present, then it may be a CPR genome”. Sounds straightforward enough. But then things are always noisy, and metagenomic bins are often incomplete and/or contaminated at least initially, hence you can’t go far with absolute presence and absence counts. Then you would have to look at ratios and percentages of those group of genes to each other, and eventually end up having to come up with biologically irrelevant cut-offs. Although we microbial ecologists are pretty awesome at not losing sleep over making meaningless cutoffs as pillars of our science, we also know one should avoid cut-offs whenever they can. One of the better ways to approach to problems of this sort is to train a ‘classifier’. Here I confess that I wrote this entire paragraph to find a way to lead you to this video: a very friendly 7 minutes to understand the concept behind classification in machine learning from Google, in case you are not familiar with the concept, and tired of not being familiar with it.
OK. I used the data you see in the anvi’o figure above to train a random forest classifier using scikit learn library. The logic is very simple: tell the computer about the distribution of single-copy genes across known CPR genomes, and ask whether it thinks an unknown genome may be coming from CPR or not. In fact, I added a learning module in anvi’o to be able to do these kinds of things in a much rapid manner in the future, so beware! I also implemented two draft scripts, anvi-script-gen-CPR-classifier for the training part, and anvi-script-predict-CPR-genomes for the prediction part.
Everything here uses anvi’o v2 branch, which is not officially released yet, but you can install and use directly from the GitHub master repository.
Now using the first programs, along with the Brown_et_al-CPR-Campbell_et_al_BSCG.txt file, I can create a classifier:
$ anvi-script-gen-CPR-classifier Brown_et_al-CPR-Campbell_et_al_BSCG.txt -o cpr-bscg.classifier
RF Train .....................................: 2391 observations with 139 features grouped into 2 classes.
Classifier output ............................: cpr-bscg.classifier
We have a classifier! So far so good. But does it classify?
An E. coli genome
Let’s start with something we all know that is not coming from CPR: E. coli (this one, to be specific).
First, the standard steps of generating an anvi’o contigs database from a given FASTA file of contigs, and populating HMM search results in it:
$ anvi-gen-contigs-database -f E_coli-K12-MG1655.fa -o E_coli-K12-MG1655.db -n 'E._coli'
$ anvi-run-hmms -c E_coli-K12-MG1655.db
Then, the moment of truth:
$ anvi-script-predict-CPR-genomes cpr-bscg.classifier -c E_coli-K12-MG1655.db
Genome in "E_coli-K12-MG1655.db"
===============================================
E_coli-K12-MG1655 ............................: NOT CPR (Confidence: 100%, Size: 4,641,652, Completion: 100%)
Phew.
But of course, this is a complete genome (as anvi’o also predicts and states it in its output: Completion: 100%), and we know genomes from CPR miss a lot of single-copy genes. Would the classifier work if it was not as complete? Here I create another FASTA file with only the first half of the genome:
$ head -n `wc -l E_coli-K12-MG1655.fa | awk '{print $1 / 2}'` E_coli-K12-MG1655.fa > E_coli-K12-MG1655-half.fa
$ wc -l E_coli-K12-MG1655.fa
58022 E_coli-K12-MG1655.fa
$ wc -l E_coli-K12-MG1655-half.fa
29011 E_coli-K12-MG1655-half.fa
Then repeat the process on this incomplete genome:
$ anvi-gen-contigs-database -f E_coli-K12-MG1655-half.fa -o E_coli-K12-MG1655-half.db -n 'Half of an E. coli'
$ anvi-run-hmms -c E_coli-K12-MG1655-half.db
$ anvi-script-predict-CPR-genomes cpr-bscg.classifier -c Genomes_NCBI/E_coli-K12-MG1655-half.db --min-percent-completion 0
Genome in "E_coli-K12-MG1655-half.db"
===============================================
E_coli-K12-MG1655-half .......................: NOT CPR (Confidence: 97%, Size: 2,239,920, Completion: 30%)
Although the genome is now only 30% complete, the classifier says it is not a CPR genome with 97% confidence.
Good job, classifier. You may have just proven yourself to be not an absolute joke. For now.
A published CPR genome
How about a known CPR genome? For this, I decided to use one of the genomes (this one, to be specific) Daan Speth and his colleagues released from a wastewater treatment system in their recent publication. Their genomes were not used in Brown et al.’s study, therefore I did not use them to train the classifier. Another group with another set of genomes; the best test case:
$ wget http://www.ncbi.nlm.nih.gov/Traces/wgs/?download=LMZU01.1.fsa_nt.gz -O LMZU01.1.fsa_nt.gz
$ gzip -d LMZU01.1.fsa_nt.gz
$ anvi-script-remove-short-contigs-from-fasta LMZU01.1.fsa_nt --simplify-names -o OP11-2.fa -l 0
$ anvi-gen-contigs-database -f OP11-2.fa -o OP11-2.db -n 'OP11'
$ anvi-run-hmms -c OP11-2.db
The moment of truth:
$ anvi-script-predict-CPR-genomes cpr-bscg.classifier -c Genomes_Speth/OP11-2.db
Genome in "OP11-2.db"
===============================================
OP11-2 .......................................: CPR (Confidence: 94%, Size: 906,719, Completion: 66%)
It classifies a genome it has never seen before as CPR. So far so good!
But then I thought that it would be more fair to classify all genomes listed in the Table 1 (I guess you already figured that I am doing these experiments as I write this post). I know that only the last five genomes in this table represent CPR genomes, and the test is to identify them properly. Just for future references, I wrote this little script to download and characterize all genomes with the accession numbers listed in Table 1:
#!/bin/bash
wget http://www.ncbi.nlm.nih.gov/Traces/wgs/?download="$1"01.1.fsa_nt.gz -O "$1"01.1.fsa_nt.gz
gzip -d "$1"01.1.fsa_nt.gz
anvi-script-remove-short-contigs-from-fasta "$1"01.1.fsa_nt --simplify-names -o $2.fa -l 0
anvi-gen-contigs-database -f $2.fa -o $2.db -n "$2"
anvi-run-hmms -c $2.db
Which I called this way for each genome from within another script:
$ ./download.sh JZEK Planctomycetes_AMX
And then classified each contigs database (the following list is in the same order with Table 1):
Planctomycetes_AMX ...........................: NOT CPR (Confidence: 92%, Size: 3,895,767, Completion: 93%)
Proteobacteria_AOB ...........................: NOT CPR (Confidence: 100%, Size: 2,610,123, Completion: 99%)
Nitrospirae_NOB ..............................: NOT CPR (Confidence: 100%, Size: 3,754,263, Completion: 91%)
Chlorobi_CHB1 ................................: NOT CPR (Confidence: 100%, Size: 2,470,583, Completion: 96%)
Chlorobi_CHB2 ................................: NOT CPR (Confidence: 98%, Size: 3,266,728, Completion: 95%)
Chlorobi_CHB3 ................................: NOT CPR (Confidence: 98%, Size: 2,549,711, Completion: 93%)
Chlorobi_CHB4 ................................: NOT CPR (Confidence: 100%, Size: 3,914,670, Completion: 96%)
Bacteroidetes_BCD1 ...........................: NOT CPR (Confidence: 100%, Size: 3,578,419, Completion: 98%)
Bacteroidetes_BCD2 ...........................: NOT CPR (Confidence: 100%, Size: 3,163,495, Completion: 98%)
Bacteroidetes_BCD3 ...........................: NOT CPR (Confidence: 100%, Size: 2,962,906, Completion: 94%)
Bacteroidetes_BCD4 ...........................: NOT CPR (Confidence: 98%, Size: 2,188,196, Completion: 86%)
Bacteroidetes_BCD5 ...........................: NOT CPR (Confidence: 100%, Size: 3,596,296, Completion: 83%)
Chloroflexi_CFX1 .............................: NOT CPR (Confidence: 100%, Size: 4,220,390, Completion: 99%)
Chloroflexi_CFX2 .............................: NOT CPR (Confidence: 100%, Size: 3,041,536, Completion: 92%)
Chloroflexi_CFX3 .............................: NOT CPR (Confidence: 100%, Size: 3,798,156, Completion: 91%)
Omnitophica_OP3 ..............................: NOT CPR (Confidence: 100%, Size: 4,052,939, Completion: 94%)
Acidobacteria_ACD ............................: NOT CPR (Confidence: 100%, Size: 2,971,784, Completion: 96%)
Armatimonadetes_ATM ..........................: NOT CPR (Confidence: 94%, Size: 2,571,275, Completion: 82%)
Parcubacteria_OD1 ............................: CPR (Confidence: 100%, Size: 904,903, Completion: 77%)
WS6_WS6-1 ....................................: CPR (Confidence: 94%, Size: 1,459,386, Completion: 78%)
WS6_WS6-2 ....................................: CPR (Confidence: 86%, Size: 1,050,508, Completion: 73%)
Microgenomates_OP11-1 ........................: CPR (Confidence: 100%, Size: 957,550, Completion: 64%)
Microgenomates_OP11-2 ........................: CPR (Confidence: 94%, Size: 906,719, Completion: 66%)
Knowing nothing about a genome except its sequence, we seem to be able to predict whether it is a member of CPR.
Screening an entire anvi’o project
Of course it is easy to screen individual genomes, or genomes well curated and finalized to perfection. But how about draft metagenomic bins? Since here in our lab we are obsessed with convenience, we made sure we will be able to screen any anvi’o bin collection rapildly with this new approach.
There is a work-in-progress project we are working on together with Ryan Bartelme and Ryan Newton. Just a week ago we had analyzed 1.4 Gb assembly generated from shotgun samples coming from a biofilter Ryan and Ryan have been studying in Milwaukee. Refining the initial CONCOCT bins with anvi’o had generated 253 bins with varius levels of completion. I thought these results would be a great test case.
This is how I screened the entire collection of bins in this analysis for CPR genomes (this is a severely cut output, I removed many many 90%+ complete and large draft genome bins from it to keep a smaller subset):
$ anvi-script-predict-CPR-genomes cpr-bscg.classifier -c CONTIGS.db -p PROFILE.db -C CONCOCT_REFINED
Contigs DB ...................................: Initialized: CONTIGS.db (v. 5)
Classifier ...................................: Initialized with 139 features grouped into 2 classes.
Num samples to classify ......................: 253.
Bins in collection "CONCOCT_REFINED"
===============================================
(...)
Bin_1_23_4 ...................................: NOT CPR (Confidence: 92%, Size: 1,140,608, Completion: 29%)
Bin_21_1 .....................................: NOT CPR (Confidence: 100%, Size: 3,090,091, Completion: 97%)
Bin_21_3 .....................................: NOT CPR (Confidence: 100%, Size: 4,696,886, Completion: 86%)
Bin_24_3 .....................................: NOT CPR (Confidence: 95%, Size: 1,612,607, Completion: 40%)
Bin_1_13 .....................................: NOT CPR (Confidence: 98%, Size: 1,983,086, Completion: 86%)
Bin_1_14 .....................................: CPR (Confidence: 95%, Size: 1,527,872, Completion: 91%)
Bin_1_18 .....................................: NOT CPR (Confidence: 98%, Size: 1,280,277, Completion: 92%)
Bin_1_4 ......................................: NOT CPR (Confidence: 100%, Size: 2,198,357, Completion: 92%)
Bin_1_3 ......................................: NOT CPR (Confidence: 100%, Size: 1,951,561, Completion: 82%)
Bin_16_17 ....................................: NOT CPR (Confidence: 92%, Size: 1,576,216, Completion: 28%)
Bin_22_7 .....................................: NOT CPR (Confidence: 100%, Size: 3,948,428, Completion: 98%)
Bin_22_5 .....................................: CPR (Confidence: 81%, Size: 703,984, Completion: 55%)
Bin_19_7 .....................................: NOT CPR (Confidence: 84%, Size: 635,704, Completion: 53%)
Bin_19_8 .....................................: NOT CPR (Confidence: 98%, Size: 1,255,111, Completion: 72%)
Bin_3_9 ......................................: CPR (Confidence: 95%, Size: 1,097,163, Completion: 14%)
Bin_8_8 ......................................: NOT CPR (Confidence: 84%, Size: 803,246, Completion: 17%)
Bin_16_13_2 ..................................: CPR (Confidence: 91%, Size: 996,627, Completion: 39%)
Bin_16_10 ....................................: NOT CPR (Confidence: 80%, Size: 645,981, Completion: 10%)
Bin_6_12 .....................................: CPR (Confidence: 88%, Size: 796,309, Completion: 32%)
Bin_16_1_6_1 .................................: NOT CPR (Confidence: 95%, Size: 1,486,735, Completion: 57%)
Bin_27_6 .....................................: NOT CPR (Confidence: 95%, Size: 1,601,108, Completion: 89%)
(...)
From 253 bins, the classifier identified 6 likely CPR bins.
To go just a little futher, I decided to focus on Bin_1_14:
Bin_1_14 .....................................: CPR (Confidence: 95%, Size: 1,527,872, Completion: 91%)
Here I ask anvi’o to bring me back all sequences from this bin that match Campbell et al. HMM profiles:
The program anvi-get-dna-sequences-for-hmm-hits is renamed to anvi-get-sequences-for-hmm-hits after anvi’o v2.1.0.
$ anvi-get-dna-sequences-for-hmm-hits -c CONTIGS.db -p PROFILE.db -C CONCOCT -b Bin_1_14 --hmm-source Campbell_et_al -o Bin_1_14.fa
Init .........................................: 71 splits in 1 bin(s)
Result .......................................: 129 genes for 1 source(s)
Output .......................................: Bin_1_14-genes.fa
The result is a FASTA file with every sequence matching to HMM profiles from Campbell et al.’s collection, and here is the RecA from that file, a phylogenetically relevant marker that may tell us what this genome bin is really from:
>RecA___7bd46ef7|bin_id:Bin_1_14|source:Campbell_et_al|e_value:6e-165|contig:k99_9268241|start:168214|stop:169330|length:1116
ATGTCGACTCTCACCGAAAAACAGAAAGCTGTAGAACTCGCGCTTTCACAAATCGAAAGAAACTTTGGCAAAGGTGCCATCATGAAACTTGGTGAATCTCAAAAGGTTGCTATCGAAACA
ATCCCTACAGGCTGTATGTCACTTGATATCGCTATCGGAGGTGGTATCCCACGTGGACGTATTATCGAAATCTTTGGACCAGAAAGCTCTGGTAAAACAACTCTCACACTTCATATCGTT
GCCCAGGCTCAAAAAATGGGCGGACAGGCAGCATTCATCGACGCTGAACACGCACTTGACCCTGAATACGCACGCAAAATCGGTGTAGATATCGATAACCTCTTAGTTTCACAGCCAGAC
AGCGGTGAACAGGCACTGGAAATTACCGAGGCGCTCGTACGTAGCAACGCAGTAGACGTAATCGTGGTGGATTCCGTAGCAGCACTCACCCCACGCGCAGAAATCGAAGGTGAAATGGGT
GACTCACACATGGGCTTACAGGCTCGTCTTATGAGCCAGGCTCTTCGTAAACTCACCAGCACCATCAGCAAATCCAAAACCACCGTCATCTTTATCAATCAGCTCCGTATGAAAATCGGT
GTAATGTTTGGTAATCCGGAAACAACAACAGGTGGTAATGCTCTTAAATTCTACGCATCTGTTCGTATGGATATCCGCAGCATCGGTAAAATCGAAGAGGGTACGGGTGAAACAAAAGAG
GTAATCGGTAATCGCGTTCGTGTAAAAGTAGTAAAAAATAAAATCGCTCCTCCATTCAAAATGGCTGAGTTCGACATCATGTATAACCGAGGTATCTCCTACACAGGTGATCTTATCGAT
CTCTCAACAAAATACGAAATCACCAGAAAGAGCGGAGCCTTCTATACATATAAAGATCTCAAACTTGGCCAAGGTCGCGAAAATGCCAAAGAGTTCCTTTCAACAAATGAAAAAGTTCTC
CGAGCTATGGAAAAAGATGTGCTTGAATATGTAGCCAACGCAAAAAAAGCTGAAGAAGTAAAAAACGAAAACTATATTCAACAGCAACAACCTCAGCAACCAAAGGCAGTTCCAGTAGCC
GCTAAAGCAGCCAAGTCAGCAAAAGATGAAGACTAA
And voilà, top hits from blastx are coming from bacteria from CPR (I removed two hypothetical hits from this list):
| Description | Max score | Total score | Query cove | E value | Ident | Accession |
|---|---|---|---|---|---|---|
| recA protein [Candidatus Peregrinibacteria bacterium GW2011_GWA2_47_7] | 406 | 406 | 88% | 2e-136 | 77% | KKU79484.1 |
| recombinase A, recombination protein RecA [Candidatus Peregrinibacteria bacterium GW2011_GWF2_39_17] | 404 | 404 | 89% | 8e-136 | 76% | KKR09265.1 |
| recombinase A, recombination protein RecA [Candidatus Peregrinibacteria bacterium GW2011_GWF2_43_17] | 390 | 390 | 86% | 3e-130 | 77% | KKT02090.1 |
| recA protein [Candidatus Peregrinibacteria bacterium GW2011_GWA2_43_8] | 390 | 390 | 86% | 3e-130 | 77% | KKT19438.1 |
| Protein RecA [Candidatus Peregrinibacteria bacterium GW2011_GWA2_38_36] | 388 | 388 | 90% | 2e-129 | 73% | KKQ71843.1 |
| Protein RecA [Candidatus Peregrinibacteria bacterium GW2011_GWA2_33_10] | 387 | 387 | 88% | 6e-129 | 75% | KKP37897.1 |
| recA protein, recombination protein RecA [Candidatus Peregrinibacteria bacterium GW2011_GWF2_38_29] | 382 | 382 | 84% | 2e-127 | 77% | KKQ68511.1 |
I could have gone further (because I am not good at knowing when to stop), but I realized I should leave the rest of the fun to Ryan Bartelme to explore these bins deeper.
I am not going into the details here right now, but if you did your metagenomic binning using another tool, you still can use anvi’o to do what is described here by generating a contigs database for your FASTA file, and using anvi-import-collection program to create a collection in it, and link contig names to bin IDs.
Final Words (tl;dr)
Tom, again, was very supportive of my decision to write this, and I thank him for making sure my expectations are set properly:
Blogs are blogs, Meren .. people don't even read papers.
I understand if you just clicked a link to get here and jumped to the end because you are as busy as the scientist next to you. So here are a couple of points I made in this post:
- The phylogeny of CPR genomes shows something extraordinary. They are truly unusual, and deserve all the buzz.
- What we have seen so far is likely the tip of the iceberg, and it is safe to assume we will see much more soon.
- It seems we can predict CPR genomes among draft genome bins from shotgun metagenomes almost instantly using single-copy genes.
- They are not specific to systems Banfield studied, and their recovery is not limited to her group. Daan Speth and his colleagues have found them. I saw on Twitter that Jen Biddle has found them, too. Although we didn’t know it before, our new classifier showed us that Ryan Bartelme and Ryan Newton’s biofilter has them as well! Now everyone knows that they do exist, more people will be looking for them. I think more light will be shed on these new clades pretty fast, and we will be humbled even further by the extent of diversity life managed to create on this planet.
These are definitely exciting times.
If you have a list of genomes you are curious to see how anvi’o would classify, but you don’t have time / interest to explore it yourelf, feel free to send me an e-mail. I will ask you to give me FASTA files, and return you back predictions.
From the discovery of a bacterium that is capable of complete nitrification to the first description of switching of viral lifestyles based on the success of the target host, microbiology has been making a great use of assembly-based shotgun metagenomics. The discovery, and the placement of some CPR genomes on the tree of life is another ground breaking discovery that took place just within the last months.
This is a good moment to stop and think about the relevance, and importance of assembly-based metagenomic approaches to the most important questions we can go after.