

Publications
Years
This page lists publications that are most reflective of our interests. For a complete list, please see Meren’s Google Scholar page.
2026
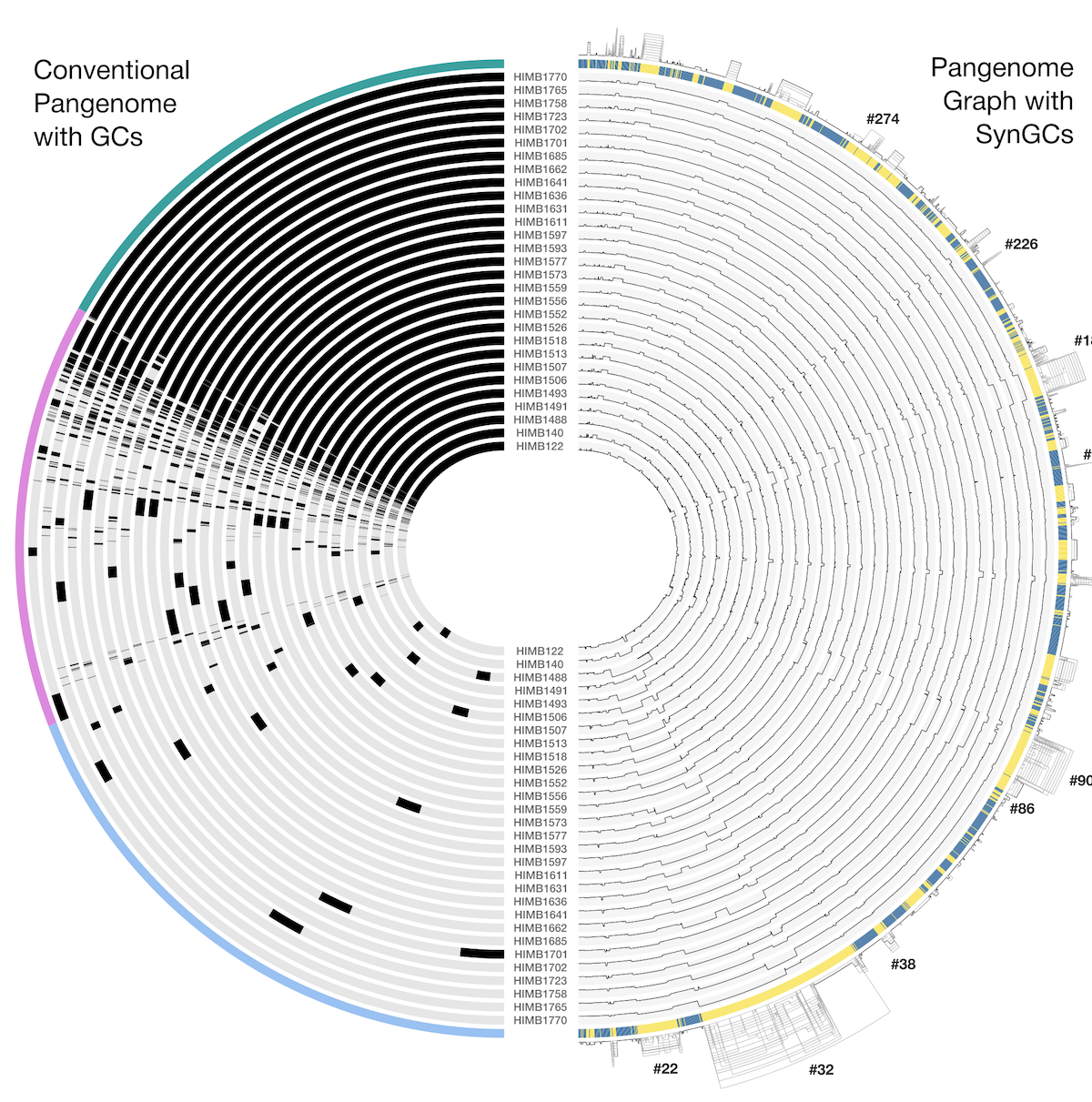
Synteny-aware microbial pangenome graphs reveal blueprints of genomic variation
 📚 bioRxiv | 🔍 Google Scholar | 🔗 doi:10.64898/2026.07.03.736256
📚 bioRxiv | 🔍 Google Scholar | 🔗 doi:10.64898/2026.07.03.736256
- Brings gene synteny into pangenomics through a novel network-pruning and graph-layout algorithm implemented in the new anvi'o programs anvi-pan-genome-graph and anvi-display-pan-graph, enabling interactive, synteny-aware visualization and quantification of backgone and variable regions in a pangenome.
- Analyzes 29 Undatipelagibacter (formerly SAR11 subclade Ia.3.VI) genomes, and reveals that genomic variability forms not a few hypervariable islands against a static backbone but a structured continuum, whose variable regions differ in scale, topology, function, and evolutionary character.
- Displays epistatic co-selection in all its glory, revaling that some evolutionary processes act on entire functional subsystems, which is a notion that have significant implications on theoretical models on genome evolution.
- A step-by-step user tutorial demonstrates how to build and explore synteny-aware pangenome graphs, and a reproducible bioinformatics workflow for our study, along with raw and intermediate data products, is available here.
- Analyzes 29 Undatipelagibacter (formerly SAR11 subclade Ia.3.VI) genomes, and reveals that genomic variability forms not a few hypervariable islands against a static backbone but a structured continuum, whose variable regions differ in scale, topology, function, and evolutionary character.
- Displays epistatic co-selection in all its glory, revaling that some evolutionary processes act on entire functional subsystems, which is a notion that have significant implications on theoretical models on genome evolution.
- A step-by-step user tutorial demonstrates how to build and explore synteny-aware pangenome graphs, and a reproducible bioinformatics workflow for our study, along with raw and intermediate data products, is available here.
Troubleshooting common errors in assemblies of long-read metagenomes
 📚 Nature Biotechnology | 🔍 Google Scholar | 🔗 doi:10.1038/s41587-025-02971-8
📚 Nature Biotechnology | 🔍 Google Scholar | 🔗 doi:10.1038/s41587-025-02971-8
- A re-evaluation of the state-of-the-art long-read assembly algorithms, including metaMDBG, hiCanu, metaFlye, and hifiasm-meta, in which we observe and describe multi-domain chimeras, prematurely circularized sequences, haplotyping errors, excessive repeats, and phantom sequences in results 😬
- A reproducible bioinformatics workflow for the study that makes use of anvi-script-find-misassemblies is available here.
- Also we have a BlueSky thread by Florian Trigodet that includes a slightly more elaborate summary if you wish to join in the conversation.
- A reproducible bioinformatics workflow for the study that makes use of anvi-script-find-misassemblies is available here.
- Also we have a BlueSky thread by Florian Trigodet that includes a slightly more elaborate summary if you wish to join in the conversation.
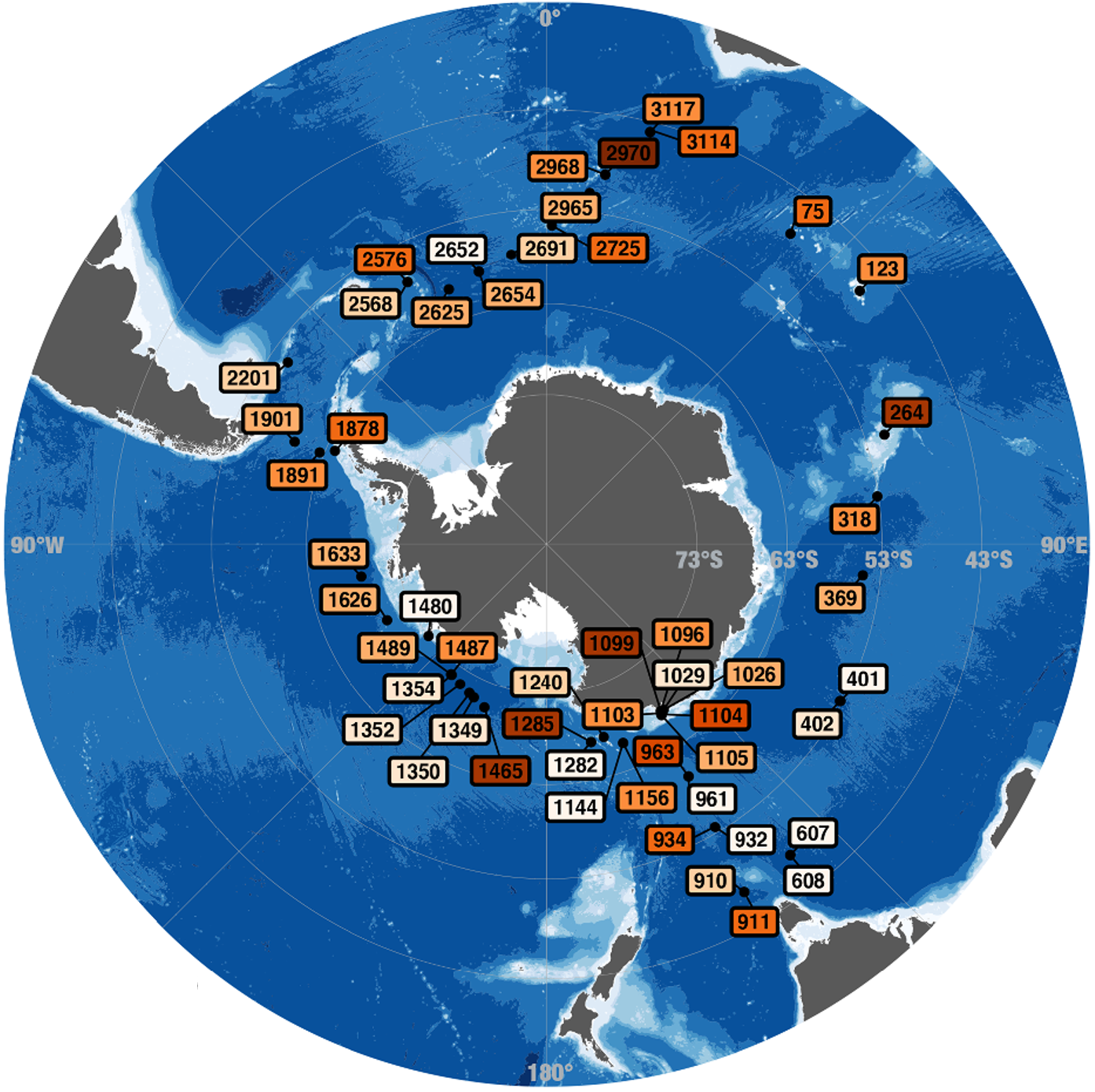
Water mass specific genes dominate the Southern Ocean microbiome
 📚 Nature Communications, 17(2025) | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-026-69584-w
📚 Nature Communications, 17(2025) | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-026-69584-w
- Introduces a Southern Ocean (SO) microbial gene catalog built from 218 metagenomes collected during the Antarctic Circumnavigation Expedition (ACE), increasing by an order of magnitude the number of SO samples ever analyzed in a metagenomics study and demonstrating that almost 40% of ACE genes lack homologs in current reference marine gene catalogs, defining the SO as a singular and largely unexplored genetic seascape.
- Shows that SO microbial gene assemblages share a common polar signature with the Arctic Ocean, yet are primarily structured by water masses at the SO scale, with biogeographical barriers at both the Sub-Antarctic Front and the Polar Front.
- Puts anvi-profile-blitz, which is a program we had implemented solely for this sutdy, to real work to profile 175 million genes across all metagenomes.
- All raw and processed data are deposited under ENA project code PRJEB83083.
- Shows that SO microbial gene assemblages share a common polar signature with the Arctic Ocean, yet are primarily structured by water masses at the SO scale, with biogeographical barriers at both the Sub-Antarctic Front and the Polar Front.
- Puts anvi-profile-blitz, which is a program we had implemented solely for this sutdy, to real work to profile 175 million genes across all metagenomes.
- All raw and processed data are deposited under ENA project code PRJEB83083.
2025
Gifting future scientists the past through well-preserved specimens of modern microbial ecosystems
 📚 Nature Communications | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-025-62138-6
📚 Nature Communications | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-025-62138-6
- A comment on why it is important to think about including 'microbial ecosystems' in modern efforts to generate archival samples for future study.
- Also makes the point that it is uncertain whether we can counteract the ongoing loss of biodiversity by resuscitating extinct organisms, and whether such organisms could survive without continuous human intervention.
- ALSO recognizes that our basic science, archival attempts, or efforts to resuscitate what is lost are unlikely to have lasting impact without transformative societal change that addresses the human drivers of biodiversity loss.
- But it conveniently passes the buck to the future generations to figure out (along with well-preserved samples that represent our time).
- Also makes the point that it is uncertain whether we can counteract the ongoing loss of biodiversity by resuscitating extinct organisms, and whether such organisms could survive without continuous human intervention.
- ALSO recognizes that our basic science, archival attempts, or efforts to resuscitate what is lost are unlikely to have lasting impact without transformative societal change that addresses the human drivers of biodiversity loss.
- But it conveniently passes the buck to the future generations to figure out (along with well-preserved samples that represent our time).
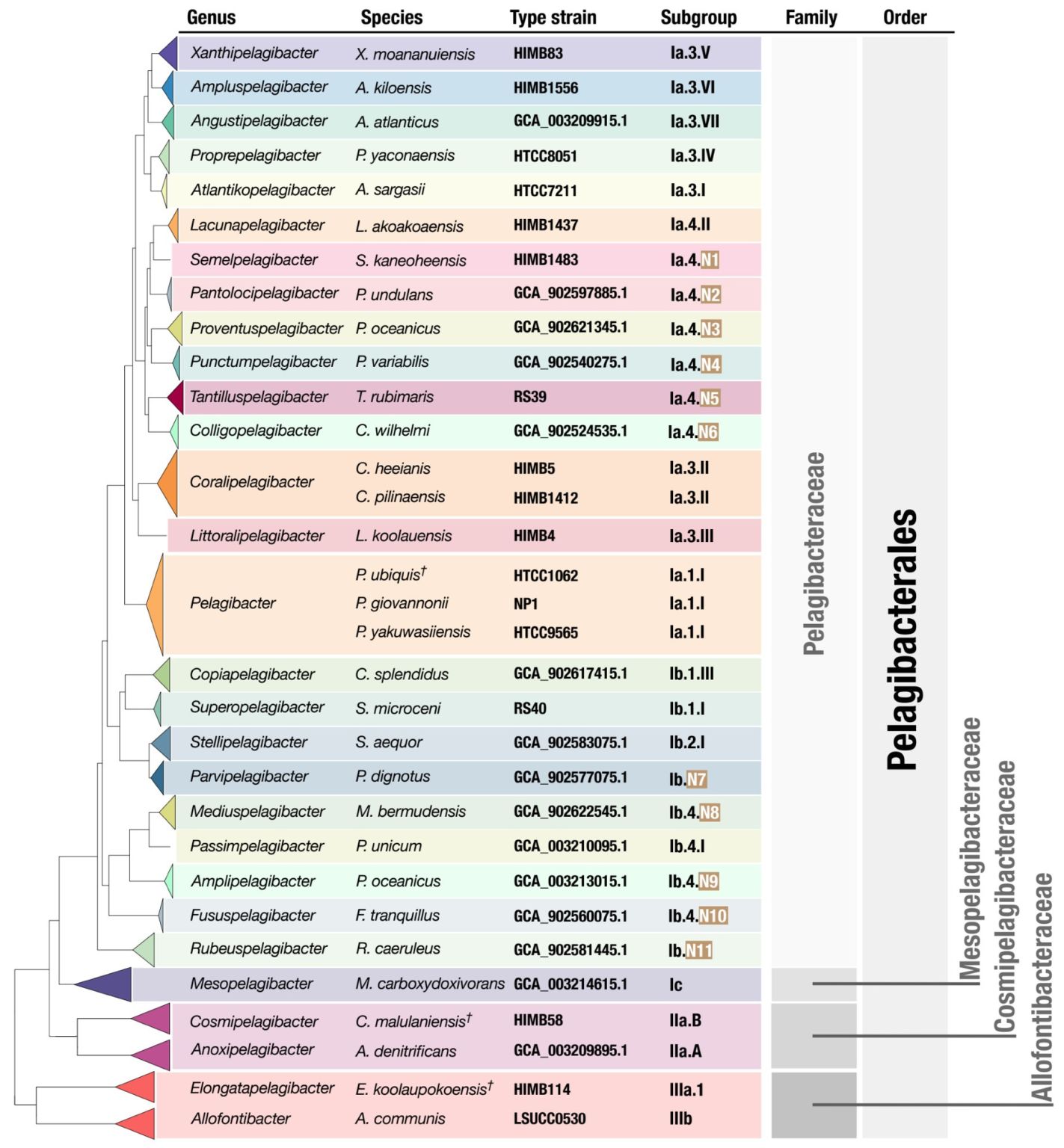
New SAR11 isolate genomes and global marine metagenomes resolve ecologically relevant units within the Pelagibacterales
 📚 Nature Communications | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-025-67043-6
📚 Nature Communications | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-025-67043-6
- Describes 81 new SAR11 cultures, which increase the total number of SAR11 cultures from the last 40 years four-fold.
- Uses the new high-quality SAR11 genomes to propose a taxonomic framework for SAR11 by introducing 29 genera within the order Pelagibacterales along with formal names for each one of them, bringing a much-needed order to SAR11 taxonomy and opens the door for community-wide discussions to resolve the nomenclature of this clade.
- You can find a little write-up by Meren on LinkedIn here.
- A reproducible bioinformatics workflow for the phylogenomics of SAR11 along with all raw and intermediate data products is available here.
- Uses the new high-quality SAR11 genomes to propose a taxonomic framework for SAR11 by introducing 29 genera within the order Pelagibacterales along with formal names for each one of them, bringing a much-needed order to SAR11 taxonomy and opens the door for community-wide discussions to resolve the nomenclature of this clade.
- You can find a little write-up by Meren on LinkedIn here.
- A reproducible bioinformatics workflow for the phylogenomics of SAR11 along with all raw and intermediate data products is available here.
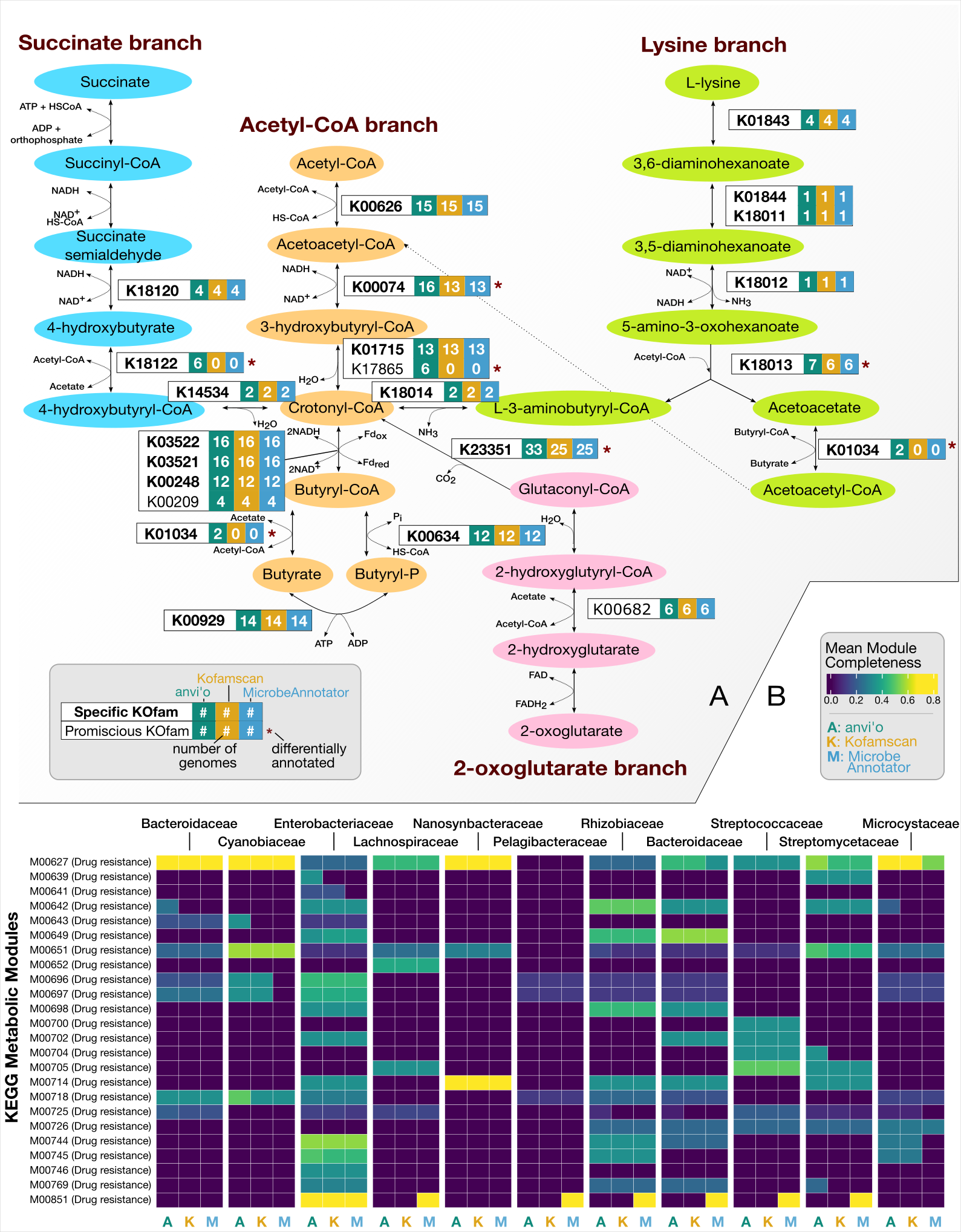
Adaptive adjustment of profile HMM significance thresholds improves functional and metabolic insights into microbial genomes
 📚 Bioinformatics Advances, 5(1):vbaf039 | 🔍 Google Scholar | 🔗 doi:10.1093/bioadv/vbaf039
📚 Bioinformatics Advances, 5(1):vbaf039 | 🔍 Google Scholar | 🔗 doi:10.1093/bioadv/vbaf039
- Demonstrates how functional annotations in and metabolic insights from microbial genomes can be substantially improved with adaptive tuning of hidden Markov model threshold while maintaining the accuracy of calls.
- An application of this strategy to annotate genes in genomes from a wide range of microbial clades showed 3% to 20% increase in functional annotation rates without having to change or update the models themselves.
- Key scripts and the workflow are available on GitHub.
- An application of this strategy to annotate genes in genomes from a wide range of microbial clades showed 3% to 20% increase in functional annotation rates without having to change or update the models themselves.
- Key scripts and the workflow are available on GitHub.
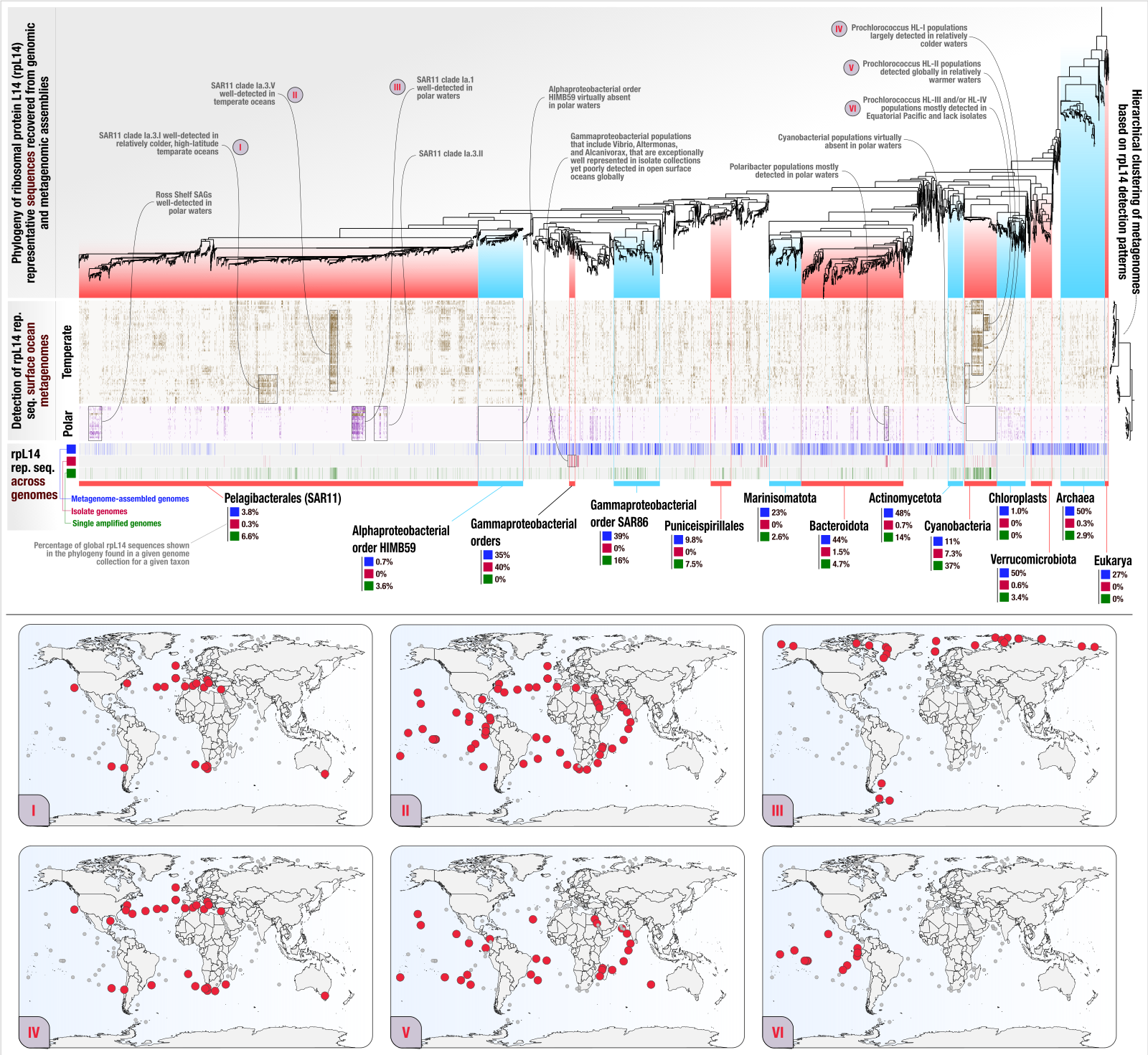
Ribosomal protein phylogeography offers quantitative insights into the efficacy of genome-resolved surveys of microbial communities
 📚 bioRxiv | 🔍 Google Scholar | 🔗 doi:10.1101/2025.01.15.633187
📚 bioRxiv | 🔍 Google Scholar | 🔗 doi:10.1101/2025.01.15.633187
- Describes the anvi'o EcoPhylo workflow, a computational workflow that gives access to the phylogeography of any gene family.
- In which we show the application of EcoPhylo to ribosomal proteins to investigate the genome recovery rates from metagenomes and demonstrate its efficacy across three biomes using genomes and metagenomes from the human gut, human oral cavity, and surface ocean.
- A reproducible bioinformatics workflow for the study is available here.
- In which we show the application of EcoPhylo to ribosomal proteins to investigate the genome recovery rates from metagenomes and demonstrate its efficacy across three biomes using genomes and metagenomes from the human gut, human oral cavity, and surface ocean.
- A reproducible bioinformatics workflow for the study is available here.
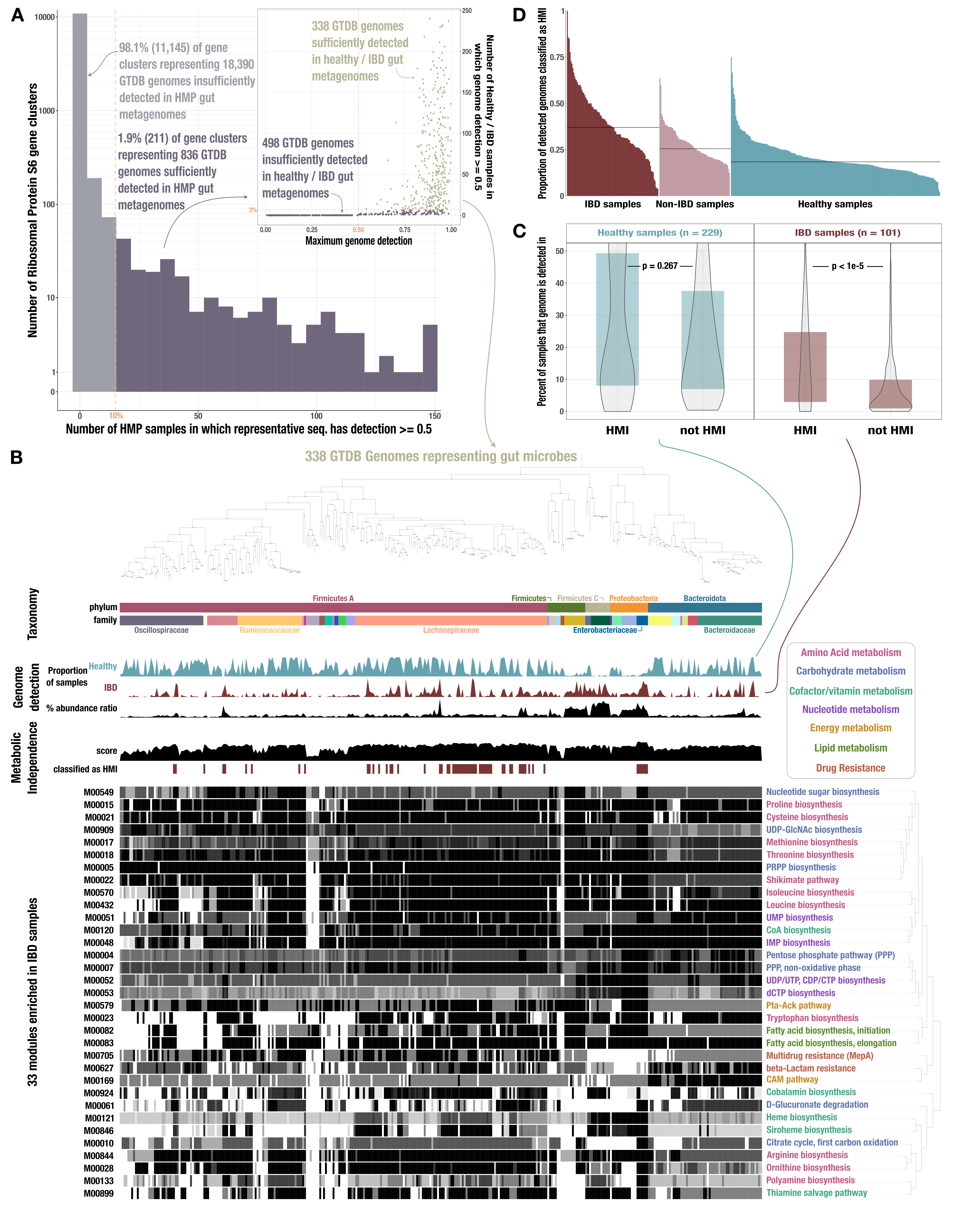
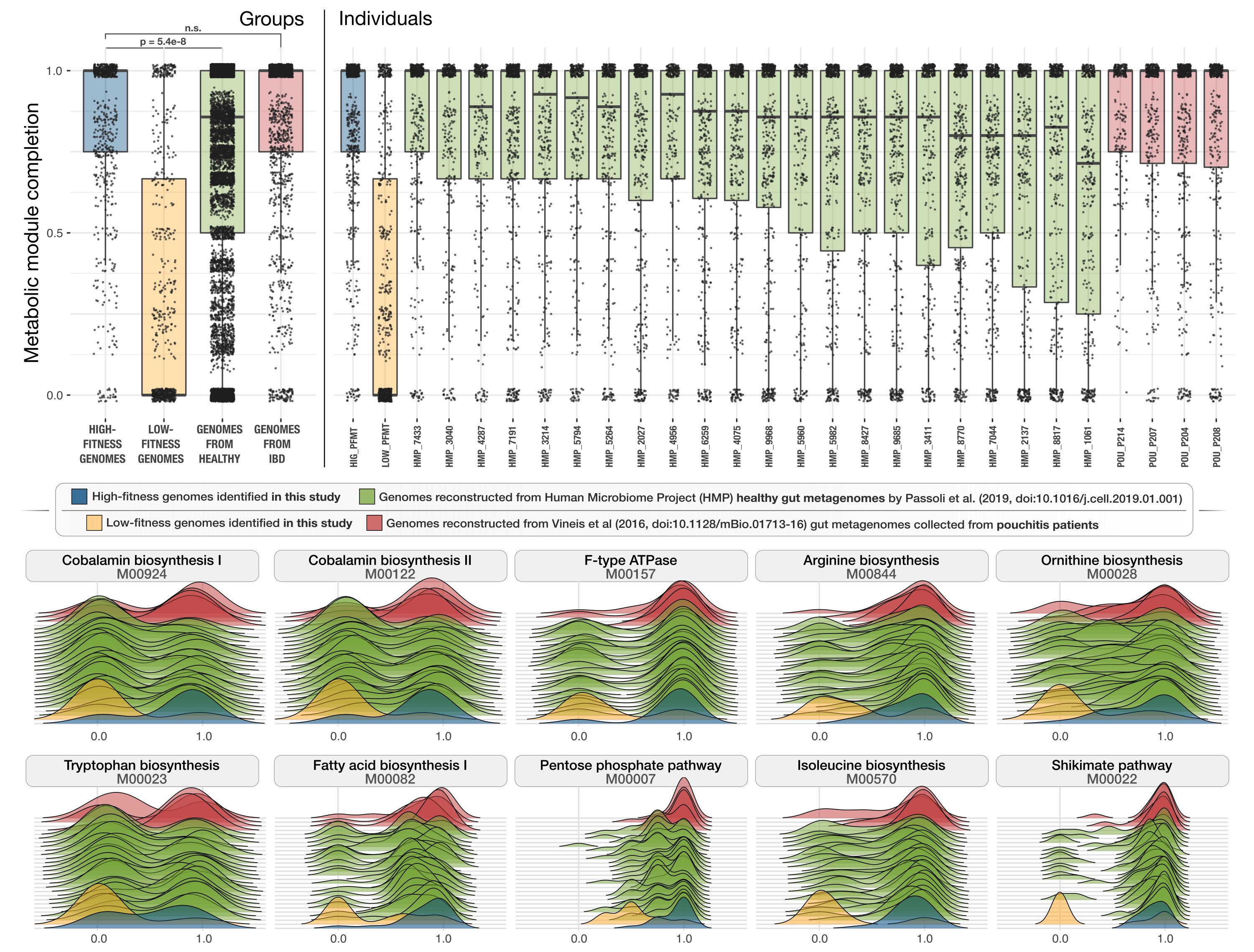
Microbes with higher metabolic independence are enriched in human gut microbiomes under stress
 📚 eLife, 12(RP89862) | 🔍 Google Scholar | 🔗 doi:10.7554/eLife.89862
📚 eLife, 12(RP89862) | 🔍 Google Scholar | 🔗 doi:10.7554/eLife.89862
- A study of microbial metabolic enrichment in human gut metagenomes that shows high metabolic independence (HMI) is a distinguishing characteristic of microbial communities associated with individuals diagnosed with IBD.
- Furthermore, it shows that the enrichment of metabolic features that are predictive of HMI and that were enriched in IBD were also enriched in gut microbiome following antibiotic treatment, suggesting that HMI is a hallmark of microbial communities in stressed gut environments.
- An insight article written by Vanessa Rossetto Marcelino, Gut Health: The value of connections, accompanies this work with additional perspectives.
- Peer reviews. Reproducible bioinformatics workflow.
- Furthermore, it shows that the enrichment of metabolic features that are predictive of HMI and that were enriched in IBD were also enriched in gut microbiome following antibiotic treatment, suggesting that HMI is a hallmark of microbial communities in stressed gut environments.
- An insight article written by Vanessa Rossetto Marcelino, Gut Health: The value of connections, accompanies this work with additional perspectives.
- Peer reviews. Reproducible bioinformatics workflow.
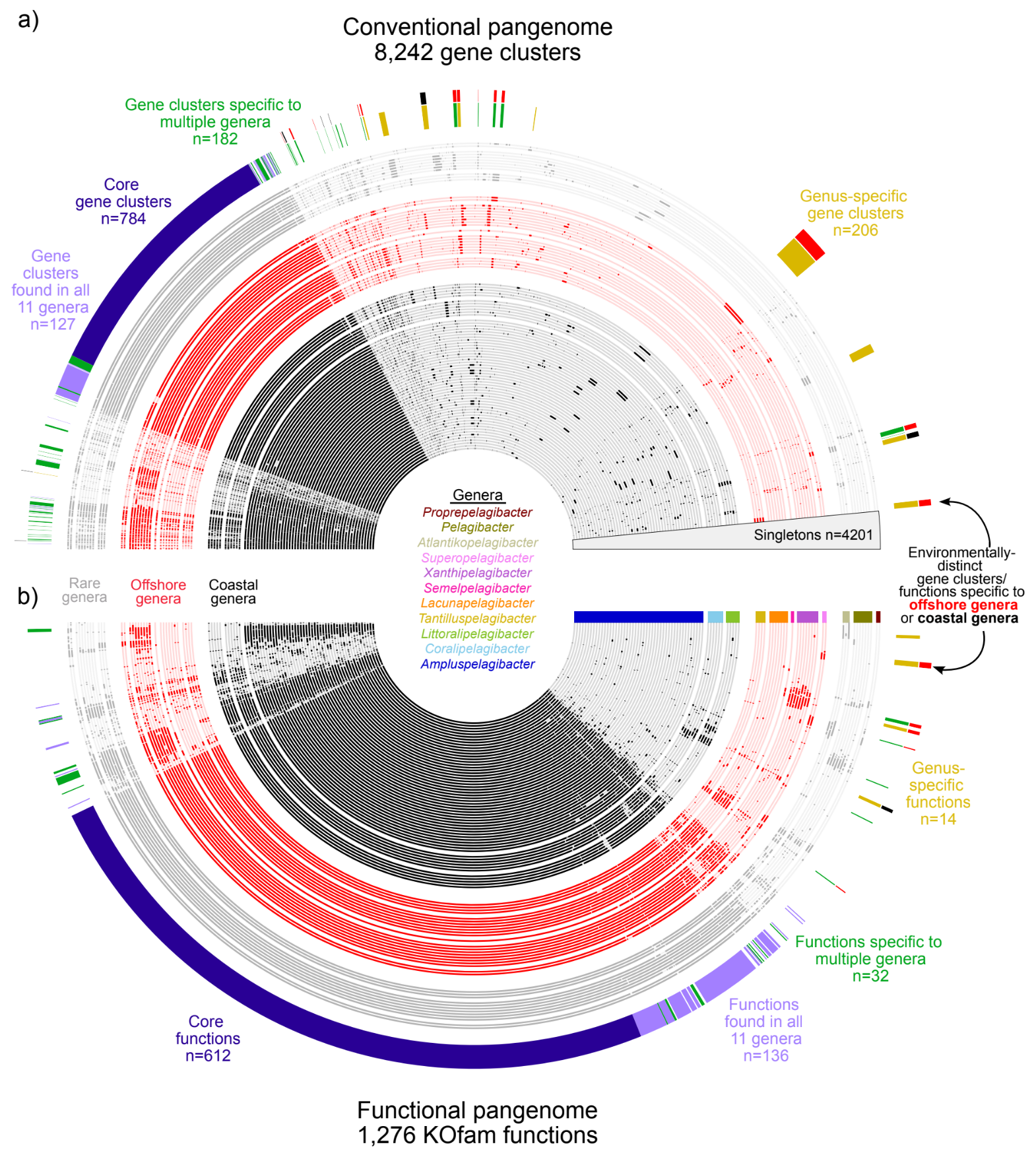
Habitat-specificity in SAR11 is associated with a few genes under high selection
 📚 The ISME Journal | 🔍 Google Scholar | 🔗 doi:10.1093/ismejo/wraf216
📚 The ISME Journal | 🔍 Google Scholar | 🔗 doi:10.1093/ismejo/wraf216
- A study that investigates the ecological differentiation within SAR11, the most abundant group of heterotrophic bacteria in the global ocean, through genomic analysis of 71 newly isolated SAR11 strains from the tropical Pacific.
- Integrates comparative genomics and environmental population genetics to show that habitat-specific distribution patterns among SAR11 subclades are linked to a handful of metabolic traits, and genes that encode them experience higher levels of selective pressures compared to most other genes in the same genome.
- Integrates comparative genomics and environmental population genetics to show that habitat-specific distribution patterns among SAR11 subclades are linked to a handful of metabolic traits, and genes that encode them experience higher levels of selective pressures compared to most other genes in the same genome.
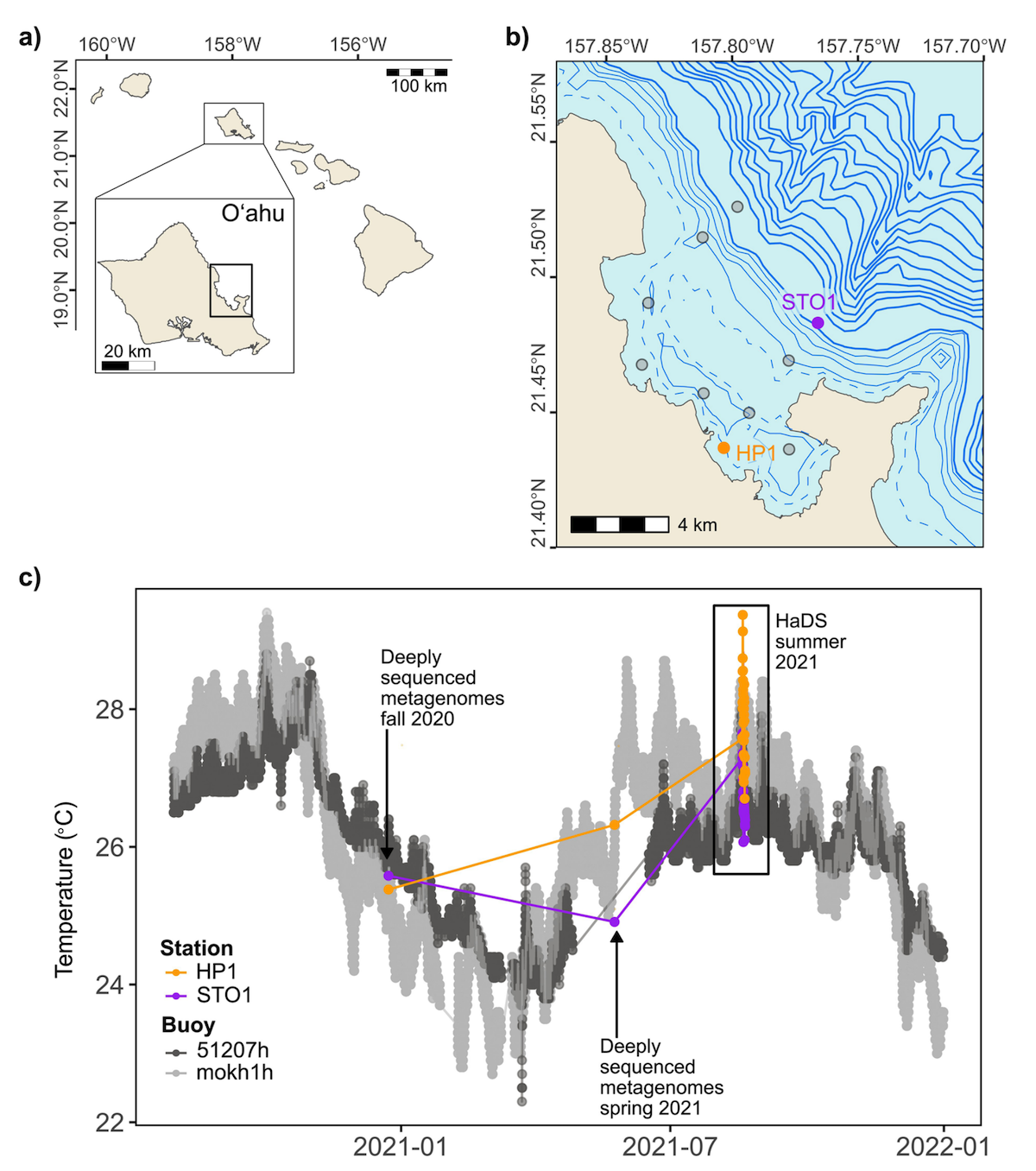
A high-resolution diel survey of surface ocean metagenomes, metatranscriptomes, and transfer RNA transcripts
 📚 Scientific Data, 12:1913 | 🔍 Google Scholar | 🔗 doi:10.1038/s41597-025-06166-3
📚 Scientific Data, 12:1913 | 🔍 Google Scholar | 🔗 doi:10.1038/s41597-025-06166-3
- Introduces the Hawaiʻi Diel Sampling (HaDS) survey, a high-resolution multi-omics dataset of surface ocean microbial communities sampled every 1.5 hours for 48 hours across two physically connected yet environmentally distinct habitats (a coastal station (HP1) in Kāneʻohe Bay and an adjacent offshore station (STO1) off Oʻahu, Hawaiʻi) to characterize microbial responses to diel changes.
- {'Altogether HaDS generated 202 intralinked sequencing products (6.62 billion paired-end short reads and 27.63 million single-end long reads)': '59 metatranscriptomes, 65 short-read metagenomes, 8 long-read metagenomes, and 66 transfer RNA (tRNA) transcripts.'}
- Provides ready-to-use data products to reduce the computational burden of reanalysis, including station-specific short- and long-read co-assemblies and 160 metagenome-assembled genomes (MAGs) from the short-read data (63 high-quality and 97 medium-quality), plus 14 high-quality and 16 medium-quality MAGs from the long-read co-assemblies.
- A reproducible bioinformatics workflow along with links to all raw and processed data products (NCBI BioProject PRJNA1201851, contigs databases, MAGs, and EcoPhylo output on FigShare) is available here.
- {'Altogether HaDS generated 202 intralinked sequencing products (6.62 billion paired-end short reads and 27.63 million single-end long reads)': '59 metatranscriptomes, 65 short-read metagenomes, 8 long-read metagenomes, and 66 transfer RNA (tRNA) transcripts.'}
- Provides ready-to-use data products to reduce the computational burden of reanalysis, including station-specific short- and long-read co-assemblies and 160 metagenome-assembled genomes (MAGs) from the short-read data (63 high-quality and 97 medium-quality), plus 14 high-quality and 16 medium-quality MAGs from the long-read co-assemblies.
- A reproducible bioinformatics workflow along with links to all raw and processed data products (NCBI BioProject PRJNA1201851, contigs databases, MAGs, and EcoPhylo output on FigShare) is available here.
2024
Modern microbiology: Embracing complexity through integration across scales
 📚 Cell, 187(19):5151-5170 | 🔍 Google Scholar | 🔗 doi:10.1016/j.cell.2024.08.028
📚 Cell, 187(19):5151-5170 | 🔍 Google Scholar | 🔗 doi:10.1016/j.cell.2024.08.028
- A review article that considers an inevitably incomplete list of emergent themes in microbiology and highlights those that are among the archetypes of its modern era.
- In which we claimed that microbiology is arguably the most important science of the mid-21st century and survived to tell the story.
- This paper was also the cover of Cell's anniversary focus issue on microbes. See this blog post by Meren to see the evolution of the cover.
- In which we claimed that microbiology is arguably the most important science of the mid-21st century and survived to tell the story.
- This paper was also the cover of Cell's anniversary focus issue on microbes. See this blog post by Meren to see the evolution of the cover.
Bioprospecting marine microbial genomes to improve biotechnology
 📚 Nature | 🔍 Google Scholar | 🔗 doi:10.1038/d41586-024-02661-6
📚 Nature | 🔍 Google Scholar | 🔗 doi:10.1038/d41586-024-02661-6
- A Nature News & Views piece to discuss the implications of the article published by Chen, Jia, Sun, and Liu et al., "Global marine microbial diversity and its potential in bioprospecting".
- We point out that the ever-increasing number of environmental genomes reported from marine systems are ready for bioprospecting efforts and to those who wish to push the boundaries of microbiology by connecting its classical and modern means.
- We point out that the ever-increasing number of environmental genomes reported from marine systems are ready for bioprospecting efforts and to those who wish to push the boundaries of microbiology by connecting its classical and modern means.
Digital Microbe: A genome-informed data integration framework for collaborative research on emerging model organisms
 📚 Scientific Data, 11(967) | 🔍 Google Scholar | 🔗 doi:10.1038/s41597-024-03778-z
📚 Scientific Data, 11(967) | 🔍 Google Scholar | 🔗 doi:10.1038/s41597-024-03778-z
- Description of a data-driven concept, 'Digital Microbe', that offers decentralized, reproducible, and interoperable means for multi-investigator teams to work on the same model organism collaboratively.
- The need and solution emerged as a function of our collaborative research in C-CoMP, the NSF-funded Science and Technology Center for Currencies of a Microbial Planet, and this publication aims to communicate our experience in this front to the community.
- The current implementation of this concept is simply a set of citable anvi'o data products, such as contigs-db and pan-db files, shared on public repositories, such as Zeonodo or FigShare.
- Reproducible bioinformatics workflows are available for the generation of the Ruegeria pomeroyi digital microbe and curation of the Altermonas pangenome as examples covered in the paper.
- The need and solution emerged as a function of our collaborative research in C-CoMP, the NSF-funded Science and Technology Center for Currencies of a Microbial Planet, and this publication aims to communicate our experience in this front to the community.
- The current implementation of this concept is simply a set of citable anvi'o data products, such as contigs-db and pan-db files, shared on public repositories, such as Zeonodo or FigShare.
- Reproducible bioinformatics workflows are available for the generation of the Ruegeria pomeroyi digital microbe and curation of the Altermonas pangenome as examples covered in the paper.
A cryptic plasmid is among the most numerous genetic elements in the human gut
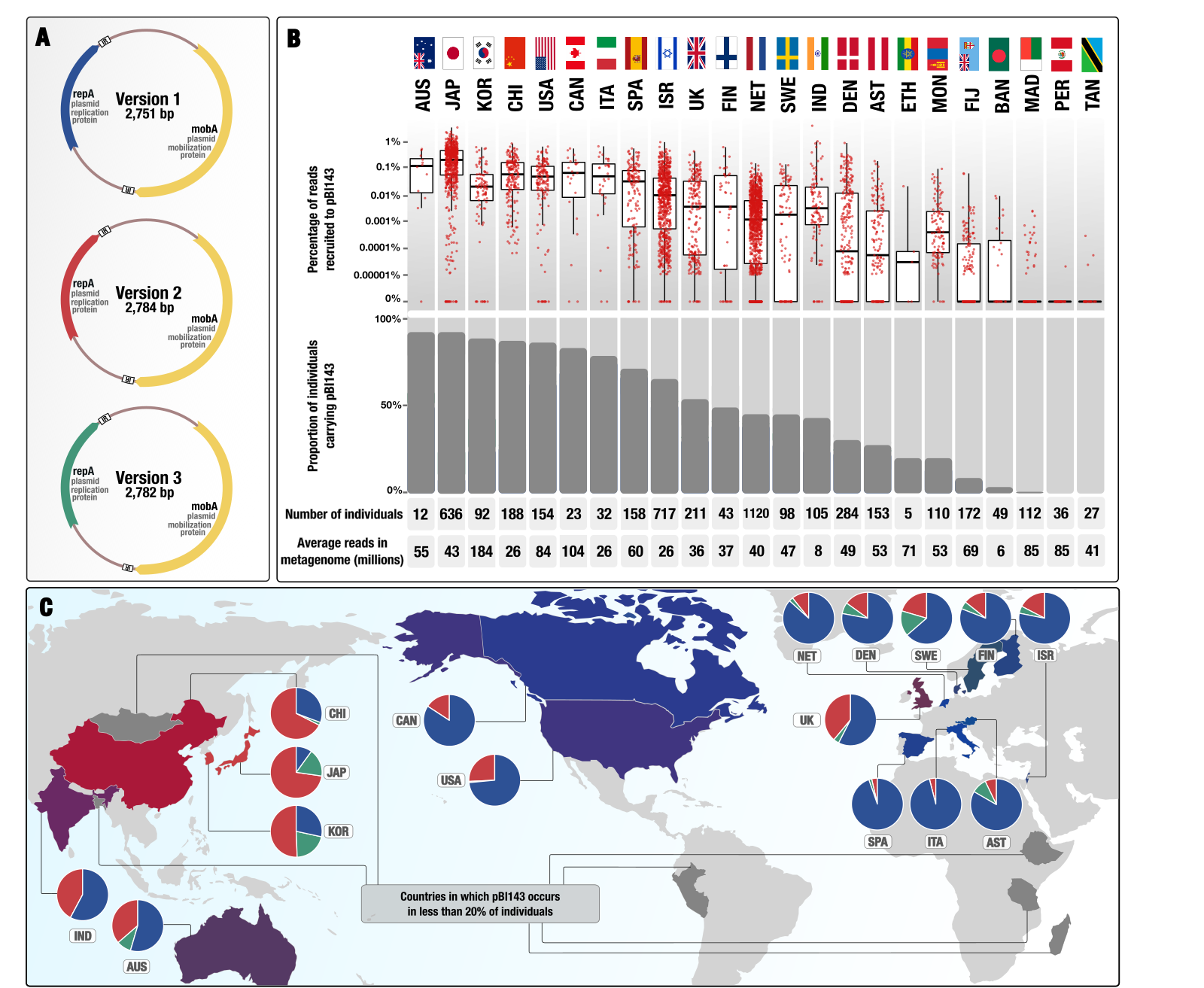 📚 Cell, 187(5):1206-1222.e16 | 🔍 Google Scholar | 🔗 doi:10.1016/j.cell.2024.01.039
📚 Cell, 187(5):1206-1222.e16 | 🔍 Google Scholar | 🔗 doi:10.1016/j.cell.2024.01.039
- A study that describes one of the most prevalent and numerous cryptic plasmid in the gut microbiomes of people who live in the industrialized world that is composed of only two genes (for its own replication and mobilization) in its native form.
- Here is a Twitter thread that explains some of the interesting aspects of pBI143 ecology as well as the practical implications having a human gut-specific and highly conserved genetic entity, copy-number of which responds to stress.
- You can find the plasmid sequences to look for pBI143 in your metagenomes, reproducible data items to re-investigate metagenomic read recruitment results, and bioinformatics workflows to elucidate population genetics of pBI143 here.
- Here is a Twitter thread that explains some of the interesting aspects of pBI143 ecology as well as the practical implications having a human gut-specific and highly conserved genetic entity, copy-number of which responds to stress.
- You can find the plasmid sequences to look for pBI143 in your metagenomes, reproducible data items to re-investigate metagenomic read recruitment results, and bioinformatics workflows to elucidate population genetics of pBI143 here.
Diverse plasmid systems and their ecology across human gut metagenomes revealed by PlasX and MobMess
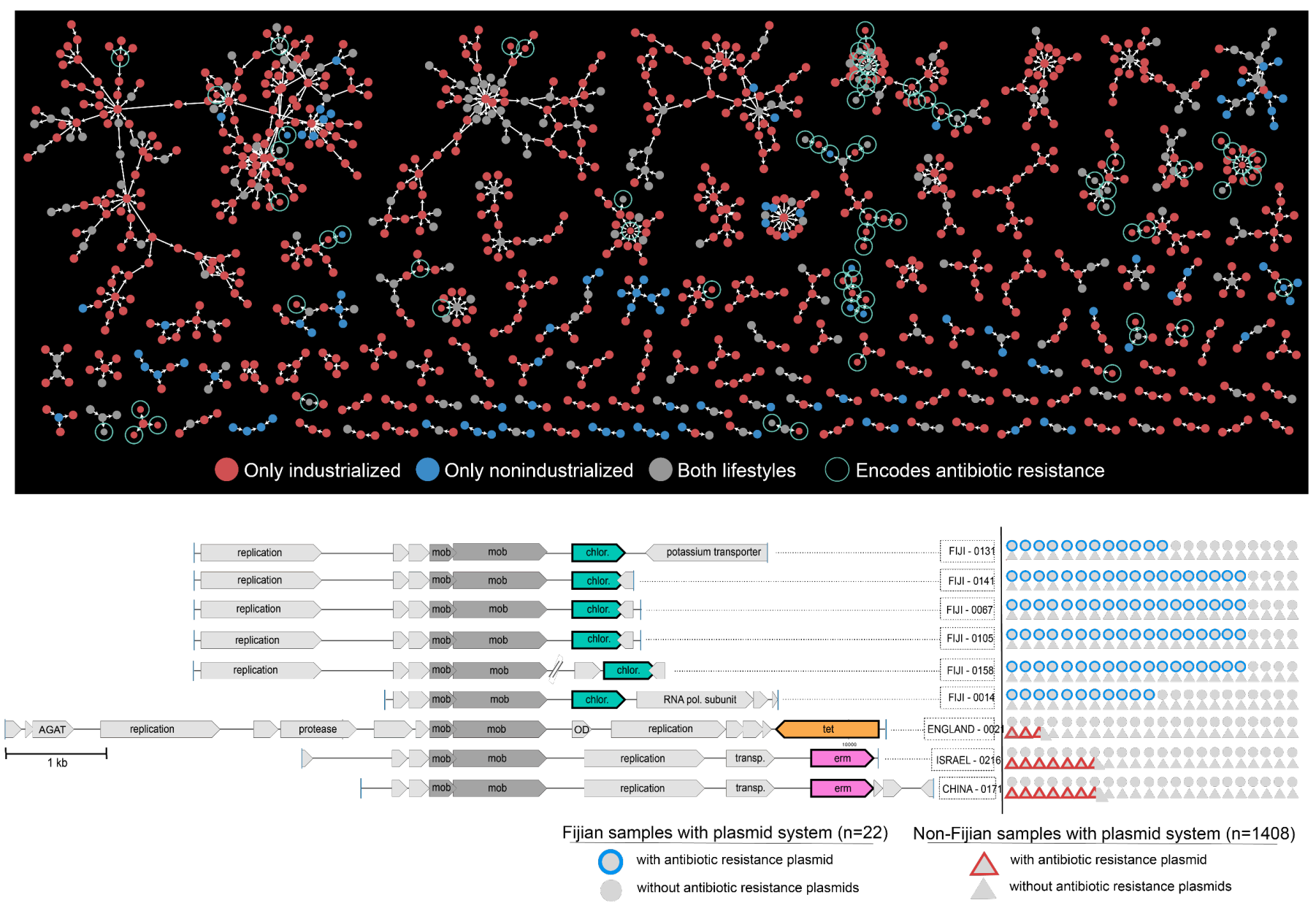 📚 Nature Microbiology, 9:830-847 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-024-01610-3
📚 Nature Microbiology, 9:830-847 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-024-01610-3
- A study that aims to shed light on the ecology and evolution of one of the most critical yet poorly studied aspects of microbial life -- naturally occurring plasmids.
- Uses state-of-the-art machine learning strategies to identify over 60,000 plasmids from human gut metagenomes, which represents a 200-fold increase in the number of known plasmids to date that were detectable in healthy humans.
- Defines hundreds of 'plasmid systems', and demonstrates that naturally occurring plasmids are not static entities, but their evolution is driven by the need to respond to the environment, and their ecology cannot be simply explained by bacterial taxonomy and distribution patterns of their putative hosts.
- Here is a Twitter thread that goes through some of the interesting aspects if this work.
- Uses state-of-the-art machine learning strategies to identify over 60,000 plasmids from human gut metagenomes, which represents a 200-fold increase in the number of known plasmids to date that were detectable in healthy humans.
- Defines hundreds of 'plasmid systems', and demonstrates that naturally occurring plasmids are not static entities, but their evolution is driven by the need to respond to the environment, and their ecology cannot be simply explained by bacterial taxonomy and distribution patterns of their putative hosts.
- Here is a Twitter thread that goes through some of the interesting aspects if this work.
Dietary- and host-derived metabolites are used by diverse gut bacteria for anaerobic respiration
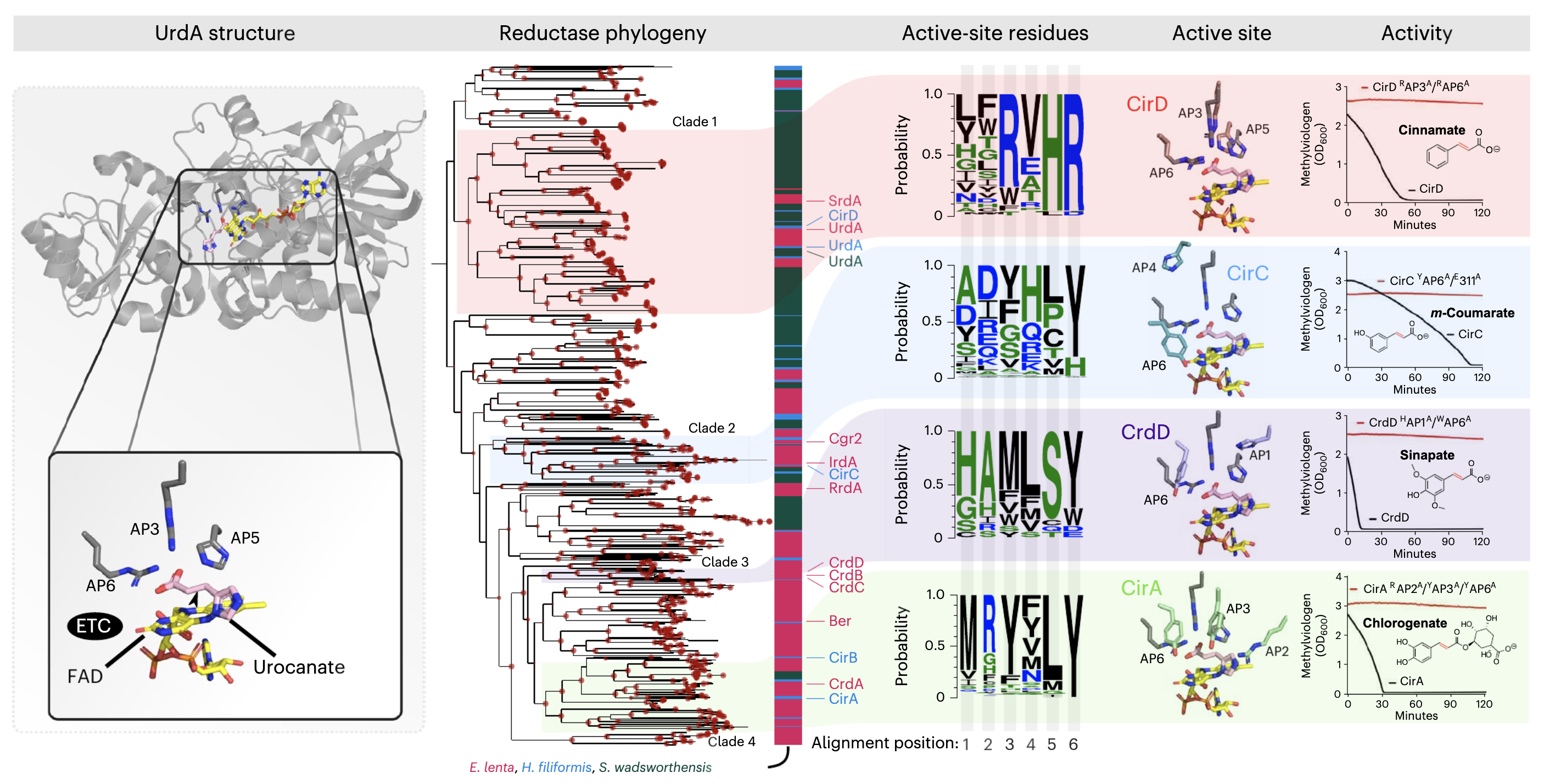 📚 Nature Microbiology, 9:55-69 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-023-01560-2
📚 Nature Microbiology, 9:55-69 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-023-01560-2
- A study that discovers three distinct taxa that encode over fifty respiratory reductases per genome that enable the use of a diverse array of metabolites as electron acceptors, establishing a respiratory strategy that utilizes the metabolite pool of the anaerobic human gut environment.
- An application of EcoPhylo to recover reductases from genomes and explain their phylogeny.
- A news article on this work from Matt Wood is also available here.
- An application of EcoPhylo to recover reductases from genomes and explain their phylogeny.
- A news article on this work from Matt Wood is also available here.
2023
Structure-informed microbial population genetics elucidate selective pressures that shape protein evolution
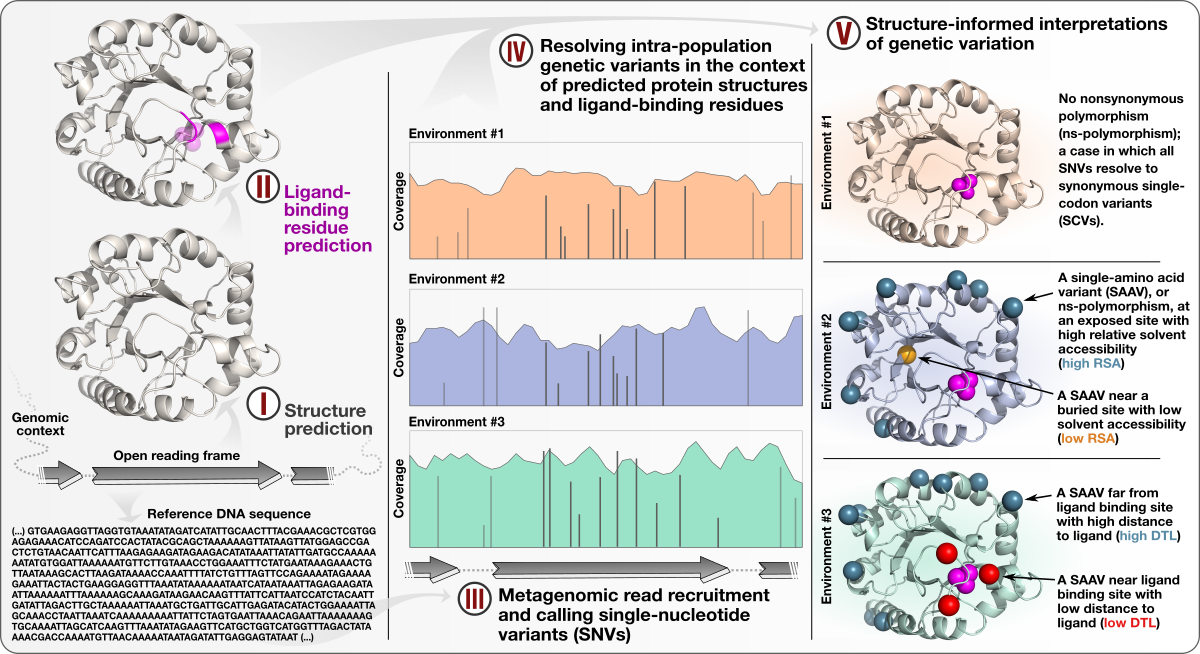 📚 Science Advances, 9(8):eabq4632 | 🔍 Google Scholar | 🔗 doi:10.1126/sciadv.abq4632
📚 Science Advances, 9(8):eabq4632 | 🔍 Google Scholar | 🔗 doi:10.1126/sciadv.abq4632
- A study that describes an approach to integrate environmental microbiology with recent advances in protein structure prediction, and illustrates the tight association between intra-population genetic variants, environmental selective pressures, and structural properties of proteins.
- Demonstrates a quantifiable link between (1) the magnitude of selective pressures over key metabolic genes (e.g., glutamine synthase of the central nitrogen metabolism), (2) the availability of key nutrients in the environment (e.g., nitrate), and (3) the maintenance of nonsynonymous variants near protein active sites.
- Comes with a reproducible bioinformatics workflow that offers detailed access to computational steps used in the study that spans from metagenomic read recruitment and profiling to the integration of environmental variants and predicted protein structures.
- Demonstrates a quantifiable link between (1) the magnitude of selective pressures over key metabolic genes (e.g., glutamine synthase of the central nitrogen metabolism), (2) the availability of key nutrients in the environment (e.g., nitrate), and (3) the maintenance of nonsynonymous variants near protein active sites.
- Comes with a reproducible bioinformatics workflow that offers detailed access to computational steps used in the study that spans from metagenomic read recruitment and profiling to the integration of environmental variants and predicted protein structures.
Metabolic independence drives gut microbial colonization and resilience in health and disease
 📚 Genome Biology, 24(78) | 🔍 Google Scholar | 🔗 doi:10.1186/s13059-023-02924-x
📚 Genome Biology, 24(78) | 🔍 Google Scholar | 🔗 doi:10.1186/s13059-023-02924-x
- A Fecal Microbiota Transplantation (FMT) study that reveals unexpected parallels between the adaptive ecological processes that shape the recipient gut microbial composition after FMT and those that influence microbial diversity in patients with Inflammatory Bowel Disease (IBD).
- Includes an observation that links the presence of superior metabolic competence in bacterial populations to their expansion in IBD.
- Here is a Twitter thread by Andrea that explains the key points of the study, and here is another one by Meren that details what are the critical learnings from it.
- Includes an observation that links the presence of superior metabolic competence in bacterial populations to their expansion in IBD.
- Here is a Twitter thread by Andrea that explains the key points of the study, and here is another one by Meren that details what are the critical learnings from it.
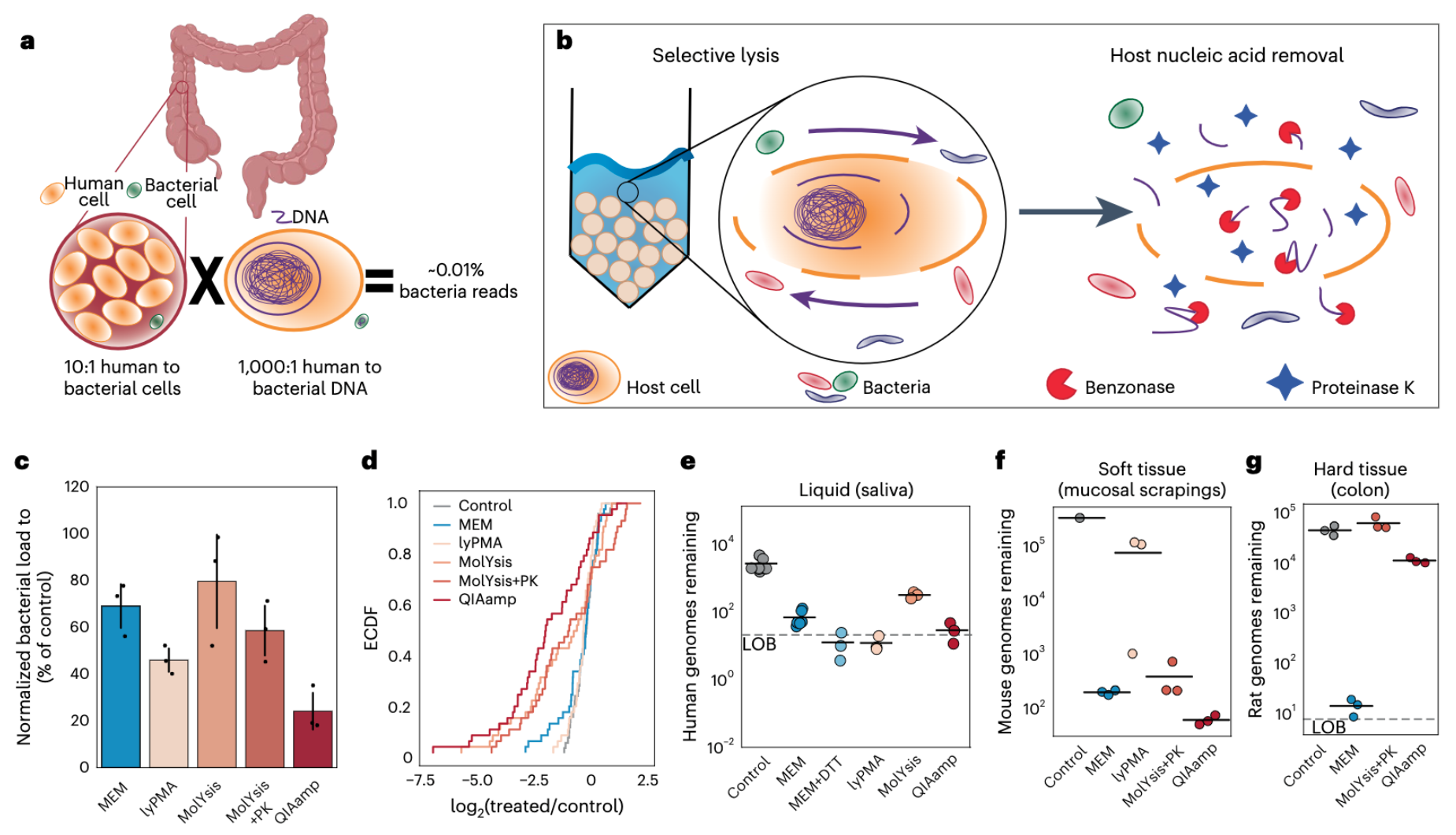
Microbial-enrichment method enables high-throughput metagenomic characterization from host-rich samples
 📚 Nature Methods | 🔍 Google Scholar | 🔗 doi:10.1038/s41592-023-02025-4
📚 Nature Methods | 🔍 Google Scholar | 🔗 doi:10.1038/s41592-023-02025-4
- A microbial-enrichment protocol that removes nucleic acids from complex samples that belong to the eukaryotic host, while not substantially perturbing the host associated microbial community composition.
- Reduce host DNA from a wide range of sample types (including human saliva, stool, intestinal scrapings, and intestinal mucosal biopsies) more than 1,000 fold with negligible changes in microbial community structures.
- Enables comprehensive surveys of microbial populations using shotgun metagenomics even in samples that have been extremely challenging to study due to extensive host contamination.
- Reduce host DNA from a wide range of sample types (including human saliva, stool, intestinal scrapings, and intestinal mucosal biopsies) more than 1,000 fold with negligible changes in microbial community structures.
- Enables comprehensive surveys of microbial populations using shotgun metagenomics even in samples that have been extremely challenging to study due to extensive host contamination.
2022
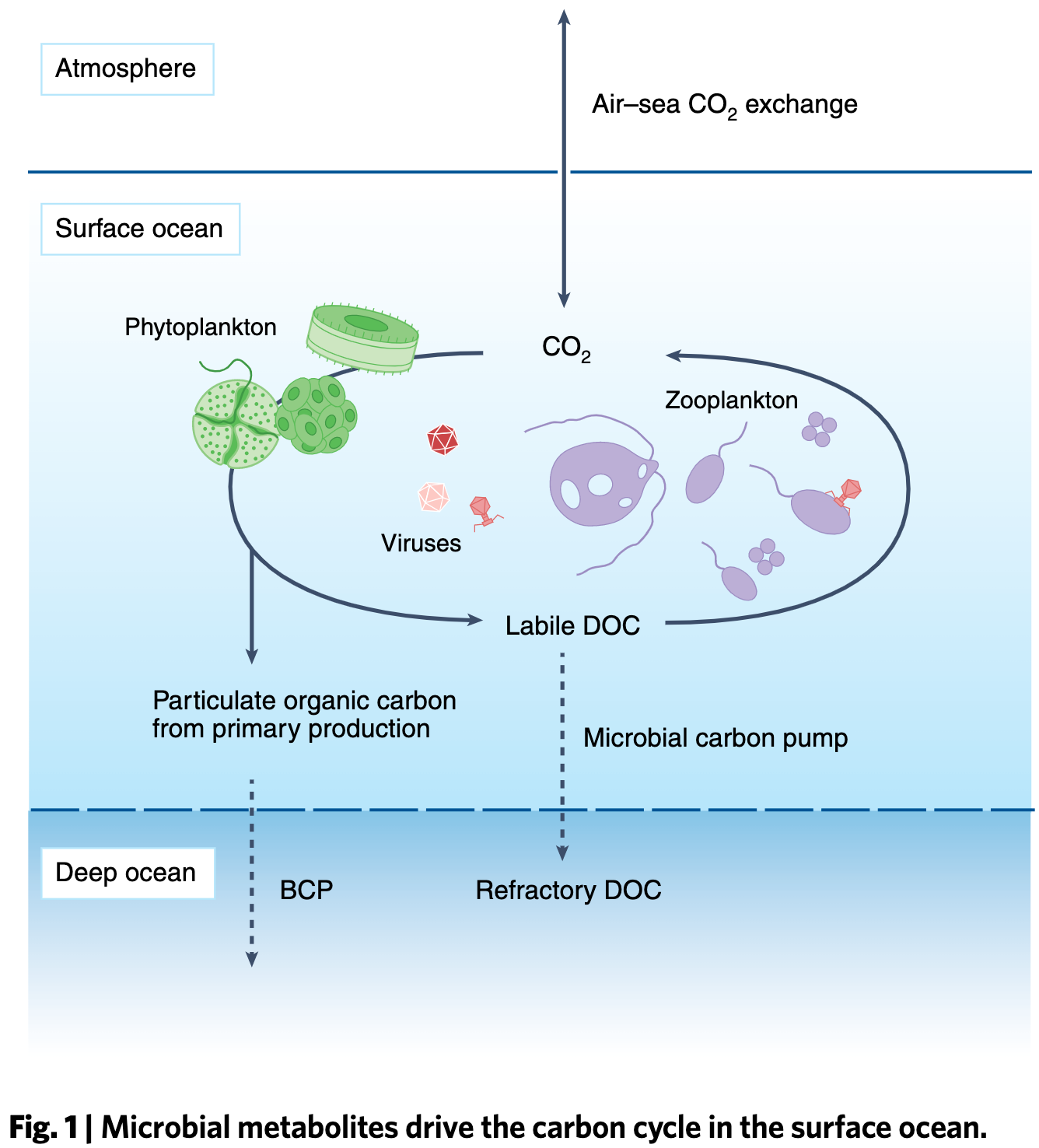
Microbial metabolites in the marine carbon cycle
 📚 Nature Microbiology, 7:508–523 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-022-01090-3
📚 Nature Microbiology, 7:508–523 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-022-01090-3
- A review that happens to be the study number 001 for the Center for Chemical Currencies of a Microbial Planet, or C-CoMP, an NSF-funded Science and Technology Center that aims to promote 'a deeper understanding of chemicals and chemical processes that underpin ocean ecosystems and the global carbon cycle by leveraging recent advances in analytical and data sciences, incorporating new ocean sampling technologies, using an open-science framework, and engaging educators and policy-makers'.
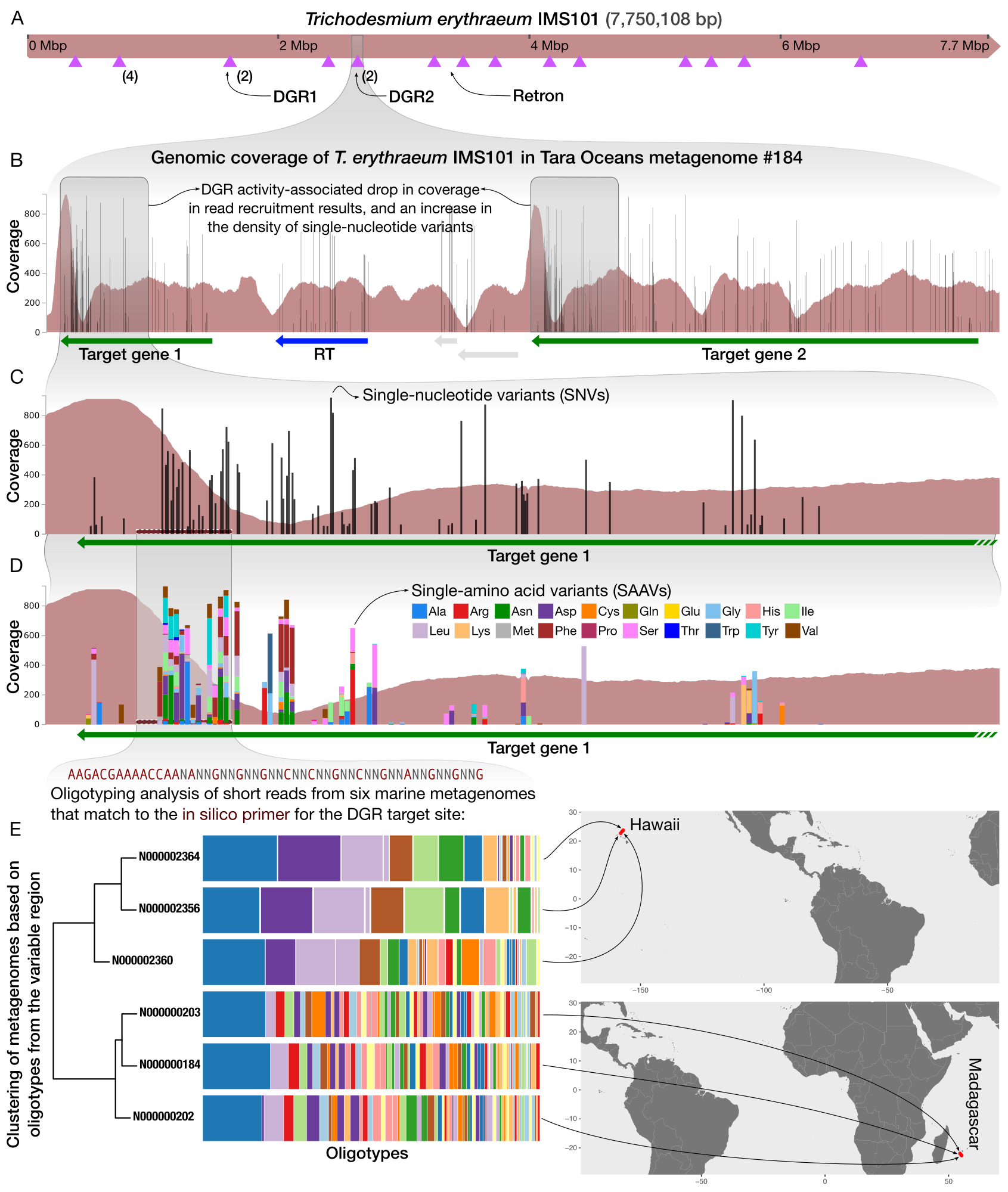
Eco-evolutionary significance of domesticated retroelements in microbial genomes
 📚 Mobile DNA, 13(6) | 🔍 Google Scholar | 🔗 doi:10.1186/s13100-022-00262-6
📚 Mobile DNA, 13(6) | 🔍 Google Scholar | 🔗 doi:10.1186/s13100-022-00262-6
- A short review on retrons and diversity-generating retro elements, some of the most beautiful and mysterious ways for life to beat the boring means of evolution and skip ahead.
- Demonstrates a workflow that gives access to the extent intra-population hypervariability of DGRs and their ecology through the analysis of metagenomes.
- A complete bioinformatics workflow that uses anvi'o and oligotyping to study DGR activity in metageomes is available here.
- Demonstrates a workflow that gives access to the extent intra-population hypervariability of DGRs and their ecology through the analysis of metagenomes.
- A complete bioinformatics workflow that uses anvi'o and oligotyping to study DGR activity in metageomes is available here.
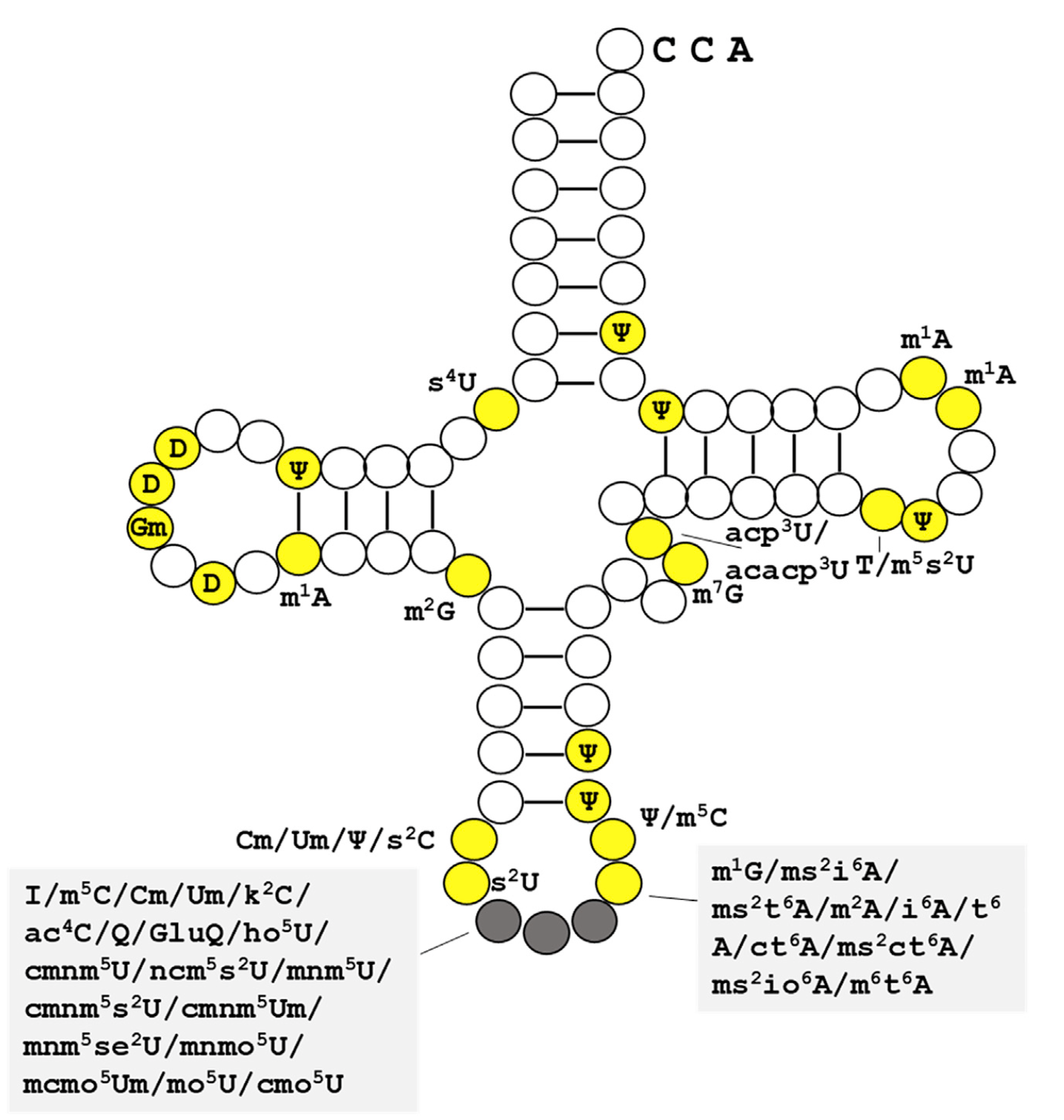
tRNA modification dynamics from individual organisms to metaepitranscriptomics of microbiomes
 📚 Molecular Cell | 🔍 Google Scholar | 🔗 doi:10.1016/j.molcel.2021.12.007
📚 Molecular Cell | 🔍 Google Scholar | 🔗 doi:10.1016/j.molcel.2021.12.007
- A review that summarizes recent advances in the studies of tRNA modification dynamics in biological processes
- Defines 'metaepitranscriptomics' as a strategy to study modification dynamics in complex environmental populations.
- Defines 'metaepitranscriptomics' as a strategy to study modification dynamics in complex environmental populations.
2021
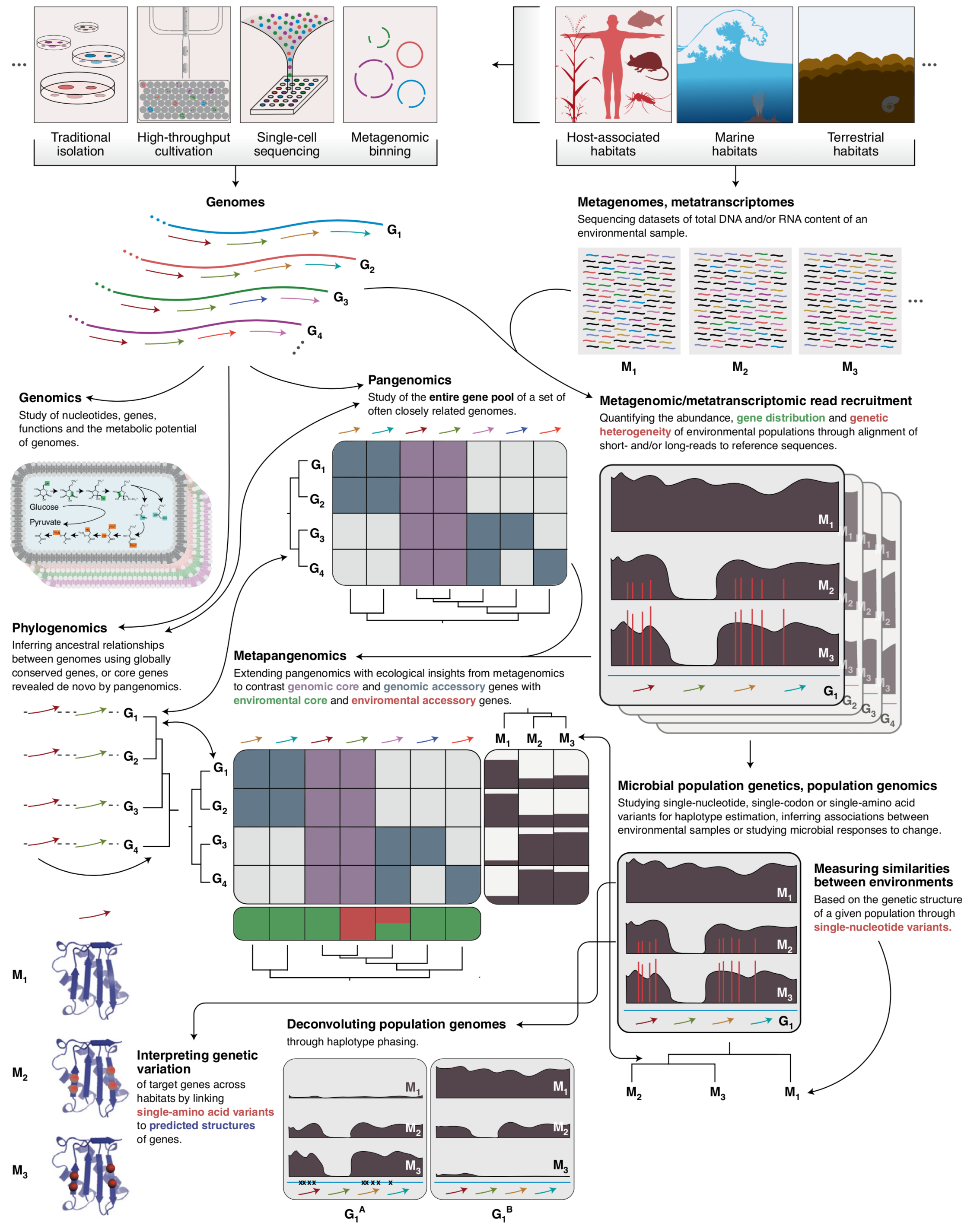
Community-led, integrated, reproducible multi-omics with anvi'o
 📚 Nature Microbiology, 6(1):186 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-020-00834-3
📚 Nature Microbiology, 6(1):186 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-020-00834-3
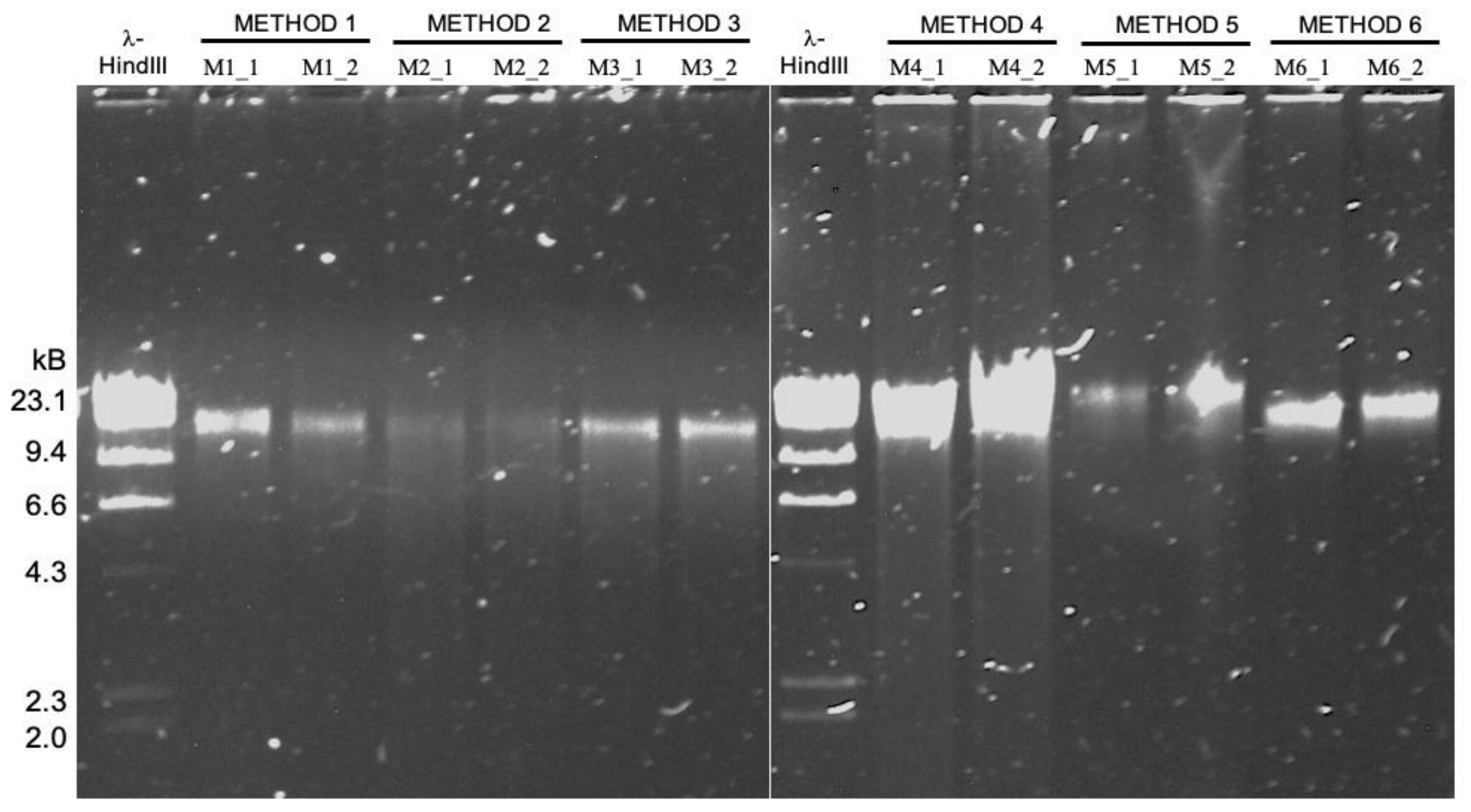
High molecular weight DNA extraction strategies for long-read sequencing of complex metagenomes
 📚 Molecular Ecology Resources, 22(5):1786-1802 | 🔍 Google Scholar | 🔗 doi:10.1111/1755-0998.13588
📚 Molecular Ecology Resources, 22(5):1786-1802 | 🔍 Google Scholar | 🔗 doi:10.1111/1755-0998.13588
- A study that benchmarks six high molecular weight DNA extraction strategies (commercially available kits, phenol-chloroform extraction, and agarose encasement followed by agarase digestion) for long-read sequencing of metagenomes with MinION.
- It turns out the protocol that works best for sequencing DNA from microbial isolates may not be the most effective method for long-read sequencing of metagenomes ¯\_(ツ)_/¯
- A reproducible bioinformatics workflow is available here. Detailed lab protocols for HMW DNA extraction methods mentioned in the study are here.
- It turns out the protocol that works best for sequencing DNA from microbial isolates may not be the most effective method for long-read sequencing of metagenomes ¯\_(ツ)_/¯
- A reproducible bioinformatics workflow is available here. Detailed lab protocols for HMW DNA extraction methods mentioned in the study are here.
2020
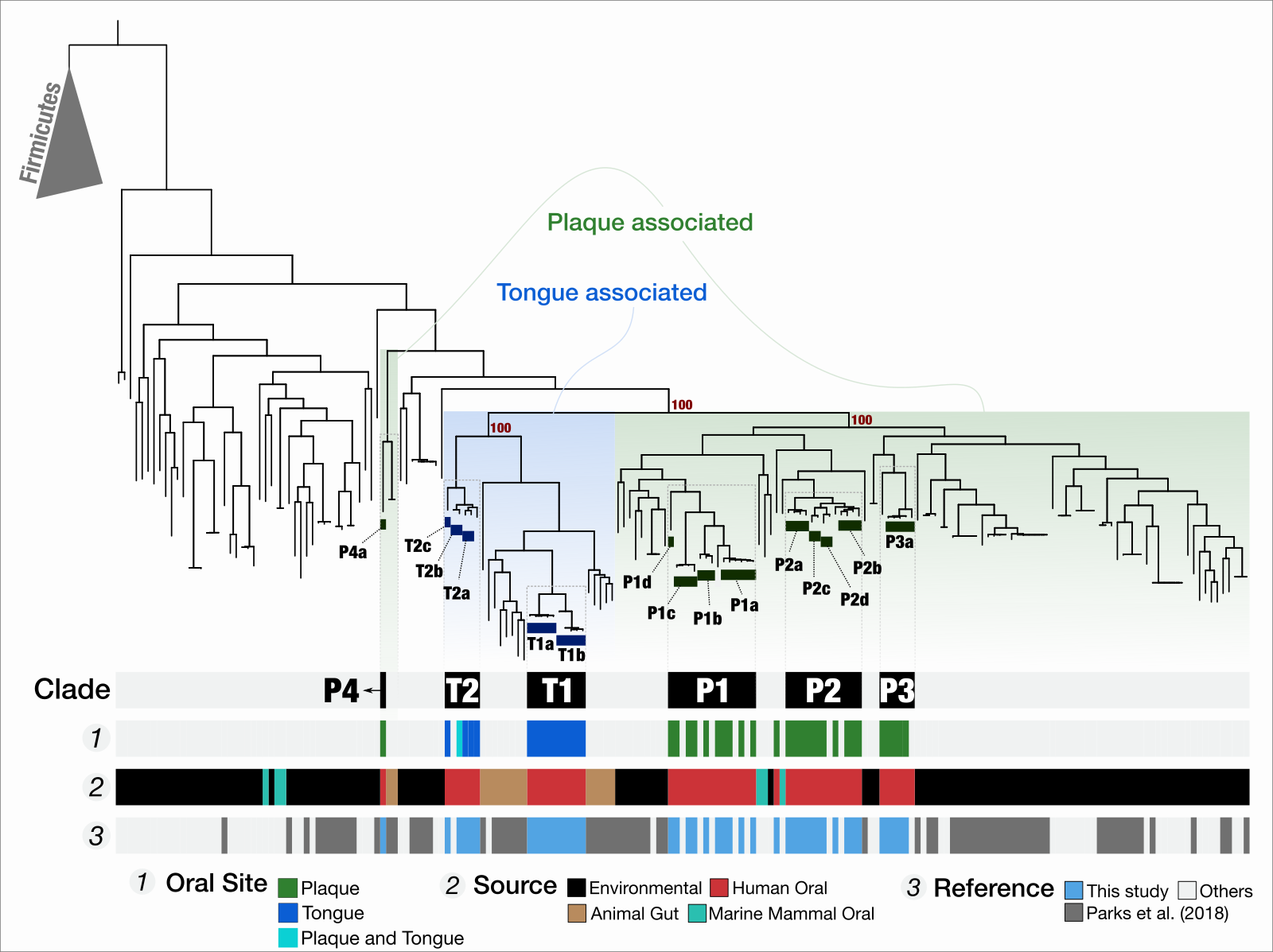
Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome
 📚 Genome Biology, 21(292) | 🔍 Google Scholar | 🔗 doi:10.1186/s13059-020-02195-w
📚 Genome Biology, 21(292) | 🔍 Google Scholar | 🔗 doi:10.1186/s13059-020-02195-w
- A multi-omics study that combines genome-resolved metagenomics, pangenomics, phylogenomics, and microbial population genetics to investigate the ecology and evolution of Saccharibacteria (TM7) in the human oral cavity, and offers a formal description of 'functional enrichment' statistic for phylogenomics and pangenomics.
- Demonstrates that TM7s split into tongue specialists and plaque specialists, and plaque TM7s group with environmental TM7s, leading to an hypothesis that the dental plaque may have served as a stepping stone for environmental microbes to adapt to host environments at least for some clades of microbes
- A news article by Alison Caldwell, PhD: Microbes in dental plaque look more like relatives in soil than those on the tongue.
- Public raw and intermediate data. Reviewer comments and responses.
- Demonstrates that TM7s split into tongue specialists and plaque specialists, and plaque TM7s group with environmental TM7s, leading to an hypothesis that the dental plaque may have served as a stepping stone for environmental microbes to adapt to host environments at least for some clades of microbes
- A news article by Alison Caldwell, PhD: Microbes in dental plaque look more like relatives in soil than those on the tongue.
- Public raw and intermediate data. Reviewer comments and responses.
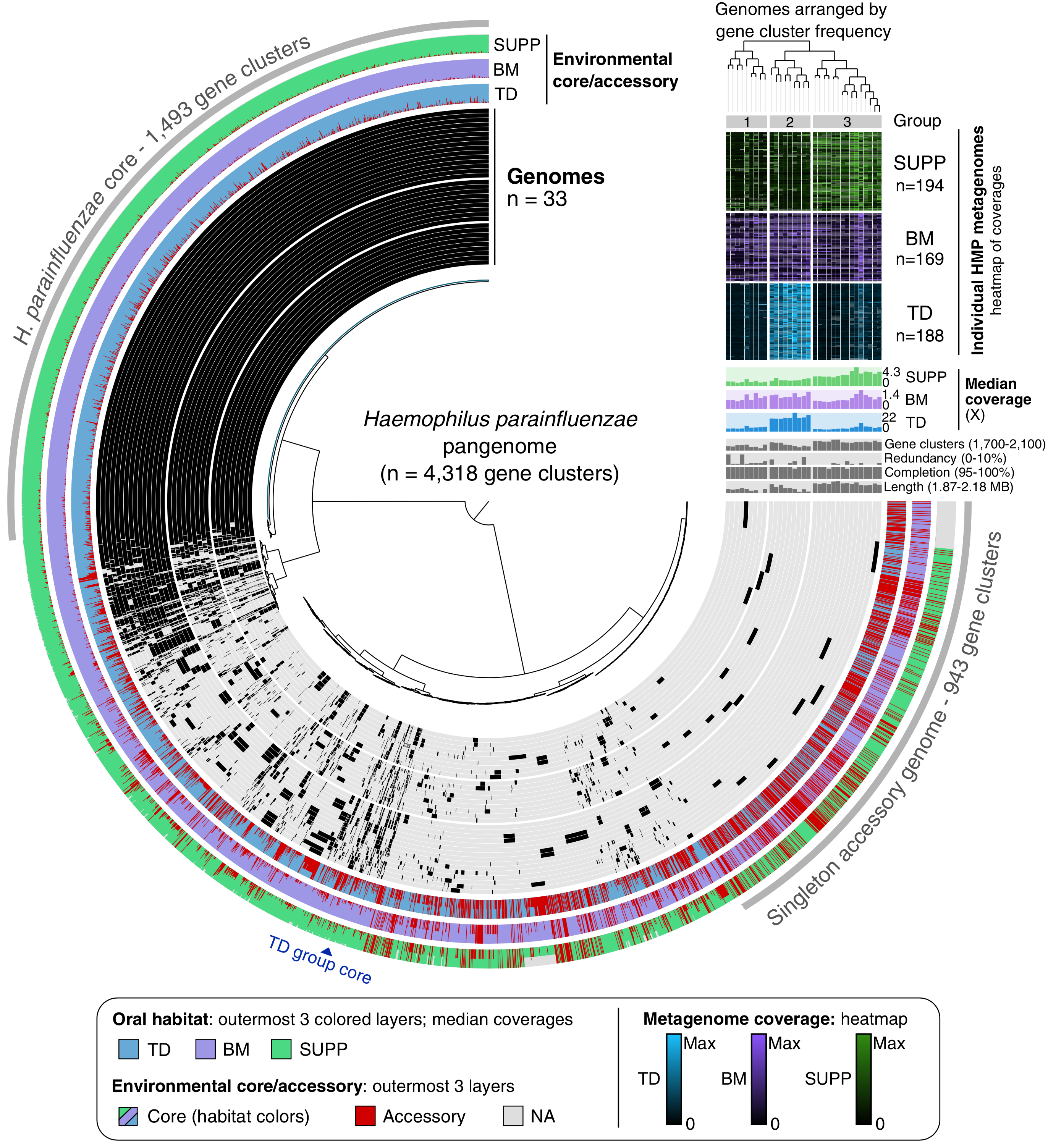
Metapangenomics of the oral microbiome provides insights into habitat adaptation and cultivar diversity
 📚 Genome Biology, 21(293) | 🔍 Google Scholar | 🔗 doi:10.1186/s13059-020-02200-2
📚 Genome Biology, 21(293) | 🔍 Google Scholar | 🔗 doi:10.1186/s13059-020-02200-2
- An application of metapangenomics that links the gene pool of two major oral microbial taxa, Haemophilus parainfluenzae and the genus Rothia, to their ecology using the Human Microbiome Project metagenomes generated from tongue, buccal mucosa, and dental plaque samples.
- Reveals that seemingly generalist organisms are composed of cryptic subpopulations with distinct ecology that is associated with only a small number of genes.
- Reviewer comments and responses. Reproducible bioinformatics workflow.
- Reveals that seemingly generalist organisms are composed of cryptic subpopulations with distinct ecology that is associated with only a small number of genes.
- Reviewer comments and responses. Reproducible bioinformatics workflow.
Droplet-based high-throughput cultivation for accurate screening of antibiotic resistant gut microbes
 📚 eLife, 9(e56998) | 🔍 Google Scholar | 🔗 doi:10.7554/eLife.56998
📚 eLife, 9(e56998) | 🔍 Google Scholar | 🔗 doi:10.7554/eLife.56998
- An aneorobic microfluidics platform for high-throughput cultivation of microbes that grows single microbial cells in millions of picoliter droplets.
- Populations of gut microbes that compete poorly in plates grow well in droplets regardless of culture media. Furthermore, taxonomic profile of droplets resembles droplets much better than plate scrapings even at the level of oligotypes.
- Demonstrates that not detecting microbes in plate-based screening of antibiotic resistance may not mean that the original sample does not contain microbes resistant to antibiotics.
- Populations of gut microbes that compete poorly in plates grow well in droplets regardless of culture media. Furthermore, taxonomic profile of droplets resembles droplets much better than plate scrapings even at the level of oligotypes.
- Demonstrates that not detecting microbes in plate-based screening of antibiotic resistance may not mean that the original sample does not contain microbes resistant to antibiotics.
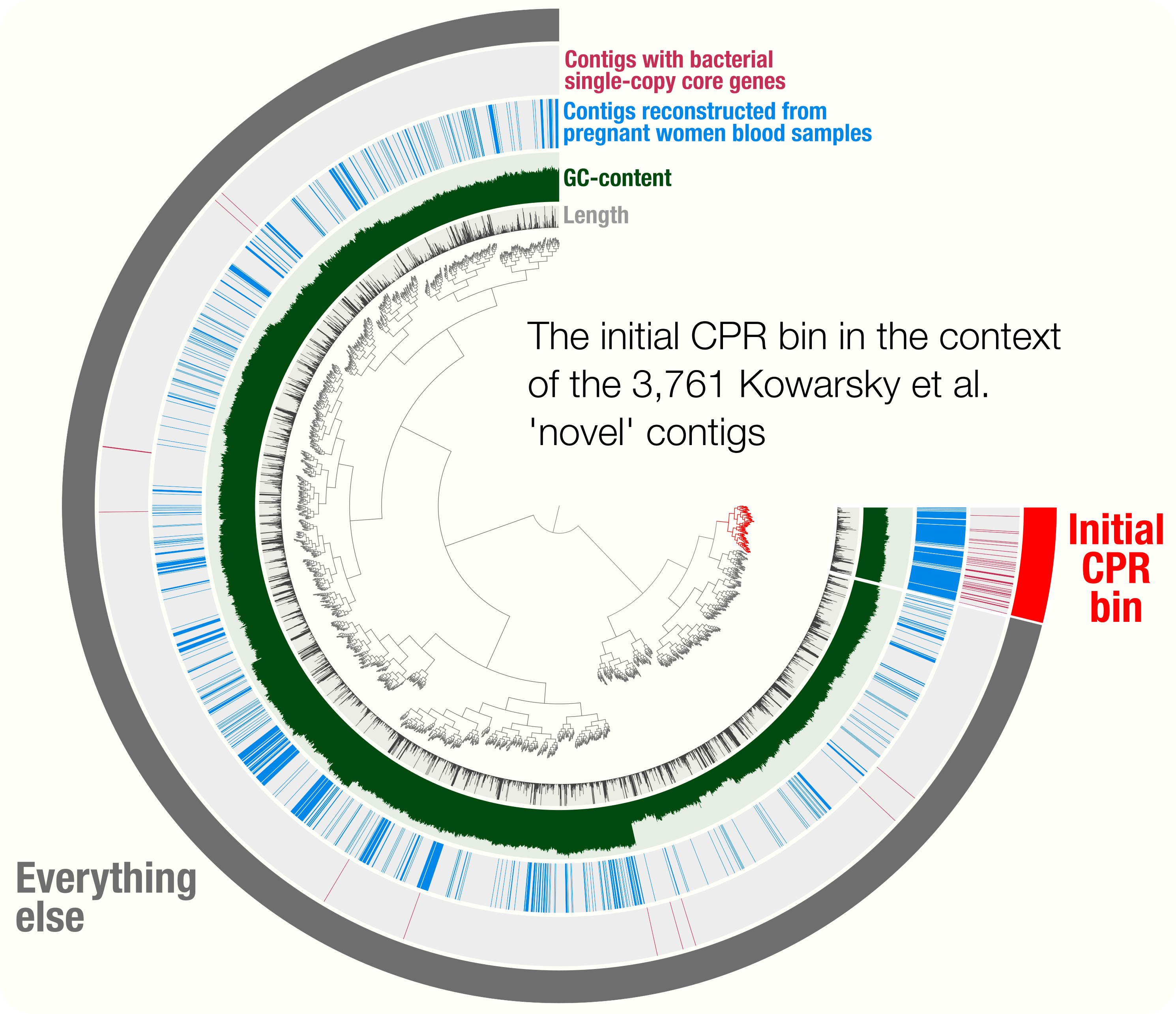
Accurate and complete genomes from metagenomes
 📚 Genome Research, 30(3):315-333 | 🔍 Google Scholar | 🔗 doi:10.1101/gr.258640.119
📚 Genome Research, 30(3):315-333 | 🔍 Google Scholar | 🔗 doi:10.1101/gr.258640.119
- A review on genome-resolved metagenomics that discusses the importance of using assembly and careful binning strategies to study metagenomes.
- Case studies include a demonstration of how single-copy core genes can fail to predict the quality of metagenome-assembled genomes, and automated strategies that yield tens of thousands of metagenome-assembled genomes will include extensive contamination.
- Promotes approaches to reconstruct 'complete' genomes from metagenomes and the use of GC skew as a metric for checking genome correctness.
- Tutorial on scaffold extension and gap closing, reproducible workflow for binning and phylogenomics of a Parcubacterium genome from human blood metagenomes.
- Case studies include a demonstration of how single-copy core genes can fail to predict the quality of metagenome-assembled genomes, and automated strategies that yield tens of thousands of metagenome-assembled genomes will include extensive contamination.
- Promotes approaches to reconstruct 'complete' genomes from metagenomes and the use of GC skew as a metric for checking genome correctness.
- Tutorial on scaffold extension and gap closing, reproducible workflow for binning and phylogenomics of a Parcubacterium genome from human blood metagenomes.
2019
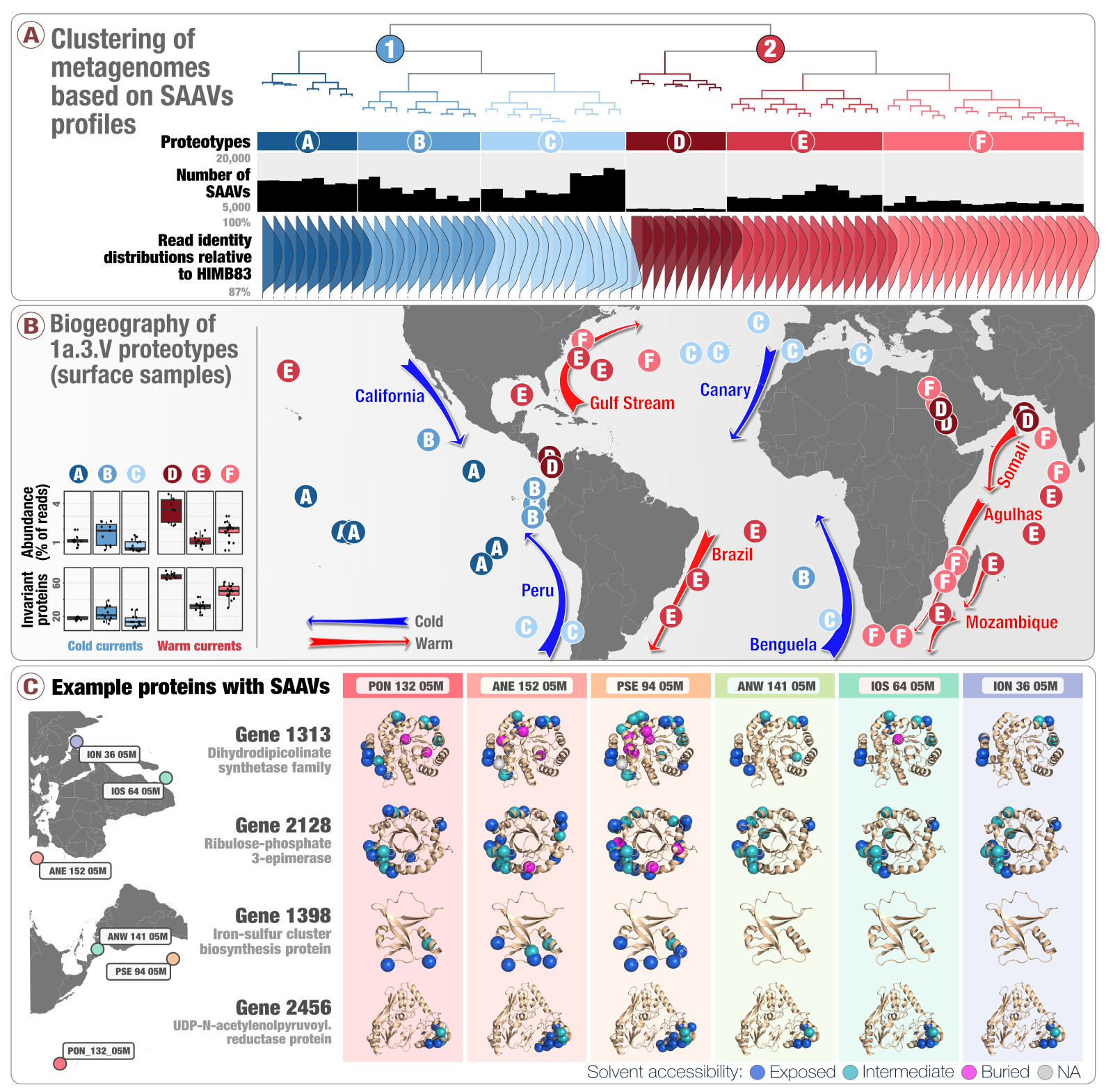
Single-amino acid variants reveal evolutionary processes that shape the biogeography of a global SAR11 subclade
 📚 eLife, 8(e46497) | 🔍 Google Scholar | 🔗 doi:10.7554/eLife.46497
📚 eLife, 8(e46497) | 🔍 Google Scholar | 🔗 doi:10.7554/eLife.46497
- A study that introduces 'single-amino acid variants' (SAAVs) and demonstrates the use of SAAVs to tease apart evolutionary processes that shape the biogeography and genomic heterogeneity within a SAR11 population through metagenomics.
- A first attempt to link population genetics and the predicted protein structures to explore in silico the intersection beetween protein biochemistry and evolutionary processes acting on an environmental microbe.
- An application of metapangenomics to define subclades of SAR11 based on gene content and ecology.
- Reproducible bioinformatics workflow is here. Reviewer criticism and our responses are also available.
- A first attempt to link population genetics and the predicted protein structures to explore in silico the intersection beetween protein biochemistry and evolutionary processes acting on an environmental microbe.
- An application of metapangenomics to define subclades of SAR11 based on gene content and ecology.
- Reproducible bioinformatics workflow is here. Reviewer criticism and our responses are also available.
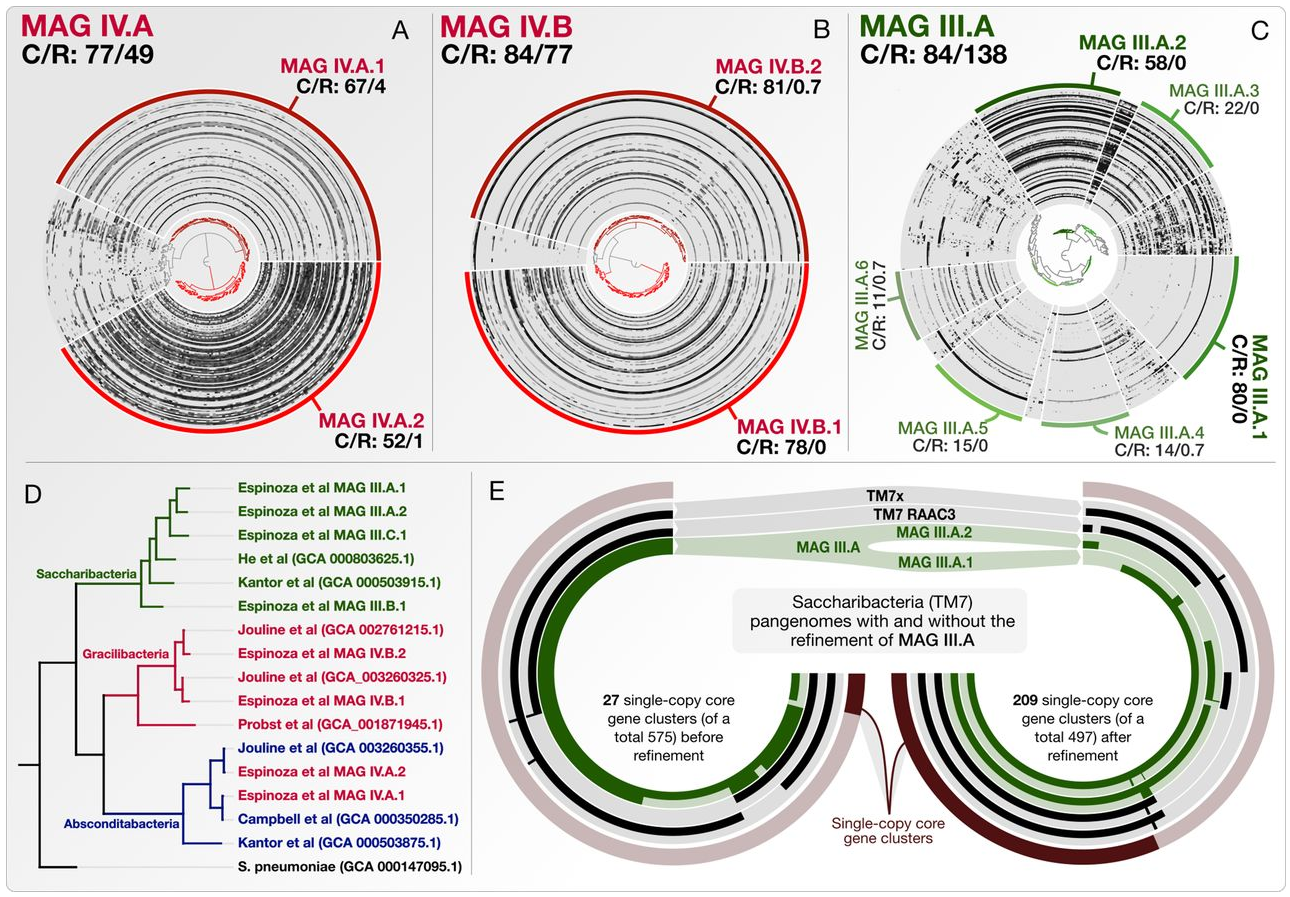
Composite Metagenome-Assembled Genomes Reduce the Quality of Public Genome Repositories
 📚 mBio, 10(3):e00725-19 | 🔍 Google Scholar | 🔗 doi:10.1128/mBio.00725-19
📚 mBio, 10(3):e00725-19 | 🔍 Google Scholar | 🔗 doi:10.1128/mBio.00725-19
- A letter that stresses that the composite metagenome-assembled genomes influence phylogenomic, pangenomic, and ecological insights (peer reviews and responses).
- A reproducible workflow to detail the steps of genome refinement and make available the refined versions of some key genomes.
- A reproducible workflow to detail the steps of genome refinement and make available the refined versions of some key genomes.
The Wolbachia mobilome in Culex pipiens includes a putative plasmid
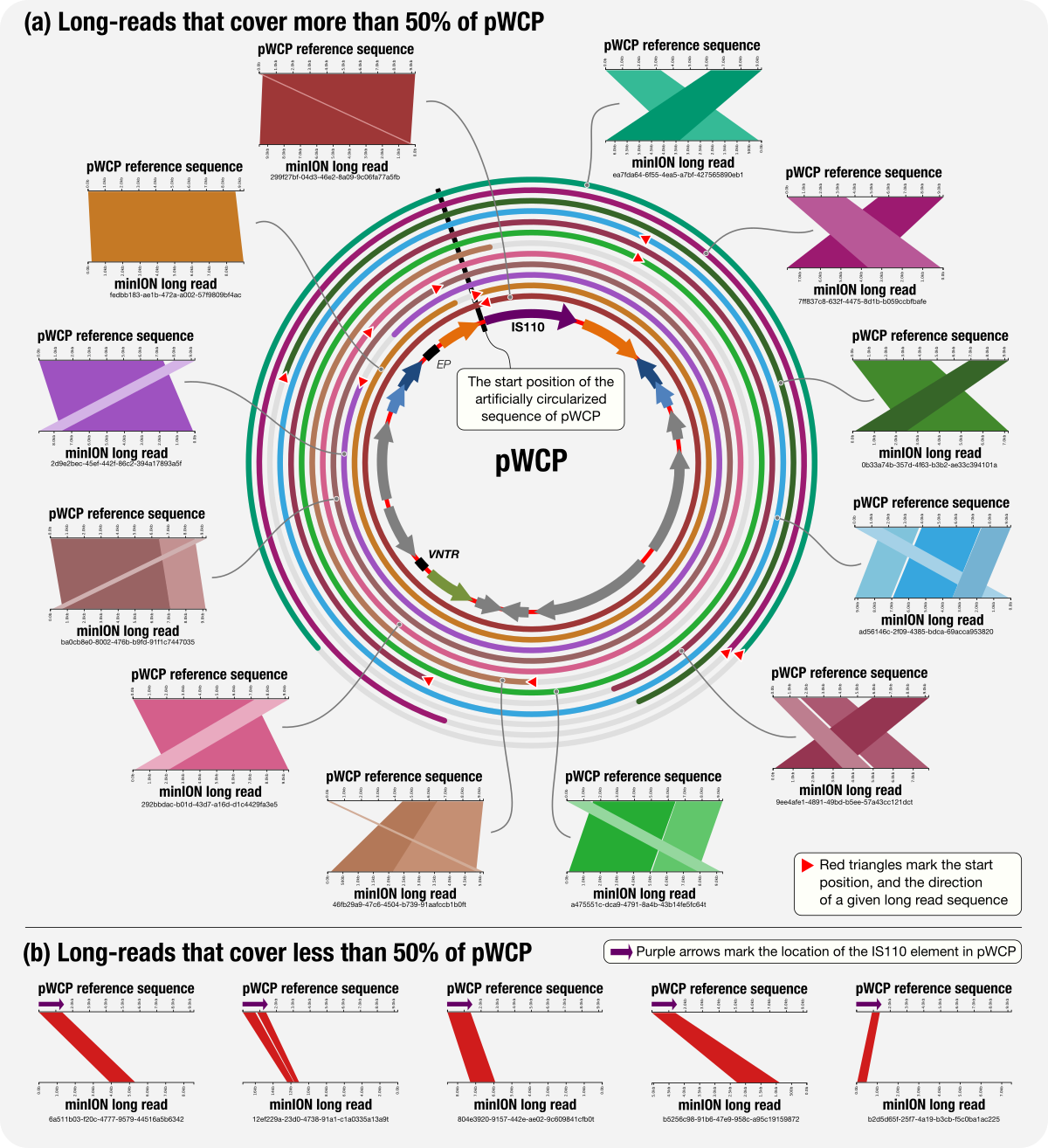 📚 Nature Communications, 10(1):1051 | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-019-08973-w
📚 Nature Communications, 10(1):1051 | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-019-08973-w
- The first report of a Wolbachia plasmid through genome-resolved metagenomics on microsurgically removed individual mosquito ovary samples (peer reviews and responses).
- Yet another application of metapangenomics and an applicatoin of minION long-read sequencing on extremely low-biomass samples.
- Reproducible bioinformatics workflow with all data items, a 'behind the paper' blog post by Julie Reveillaud, and a press release from the Marine Biological Laboratory.
- Yet another application of metapangenomics and an applicatoin of minION long-read sequencing on extremely low-biomass samples.
- Reproducible bioinformatics workflow with all data items, a 'behind the paper' blog post by Julie Reveillaud, and a press release from the Marine Biological Laboratory.
Genome-resolved insights into a novel Spiroplasma symbiont of the Wheat Stem Sawfly (Cephus cinctus)
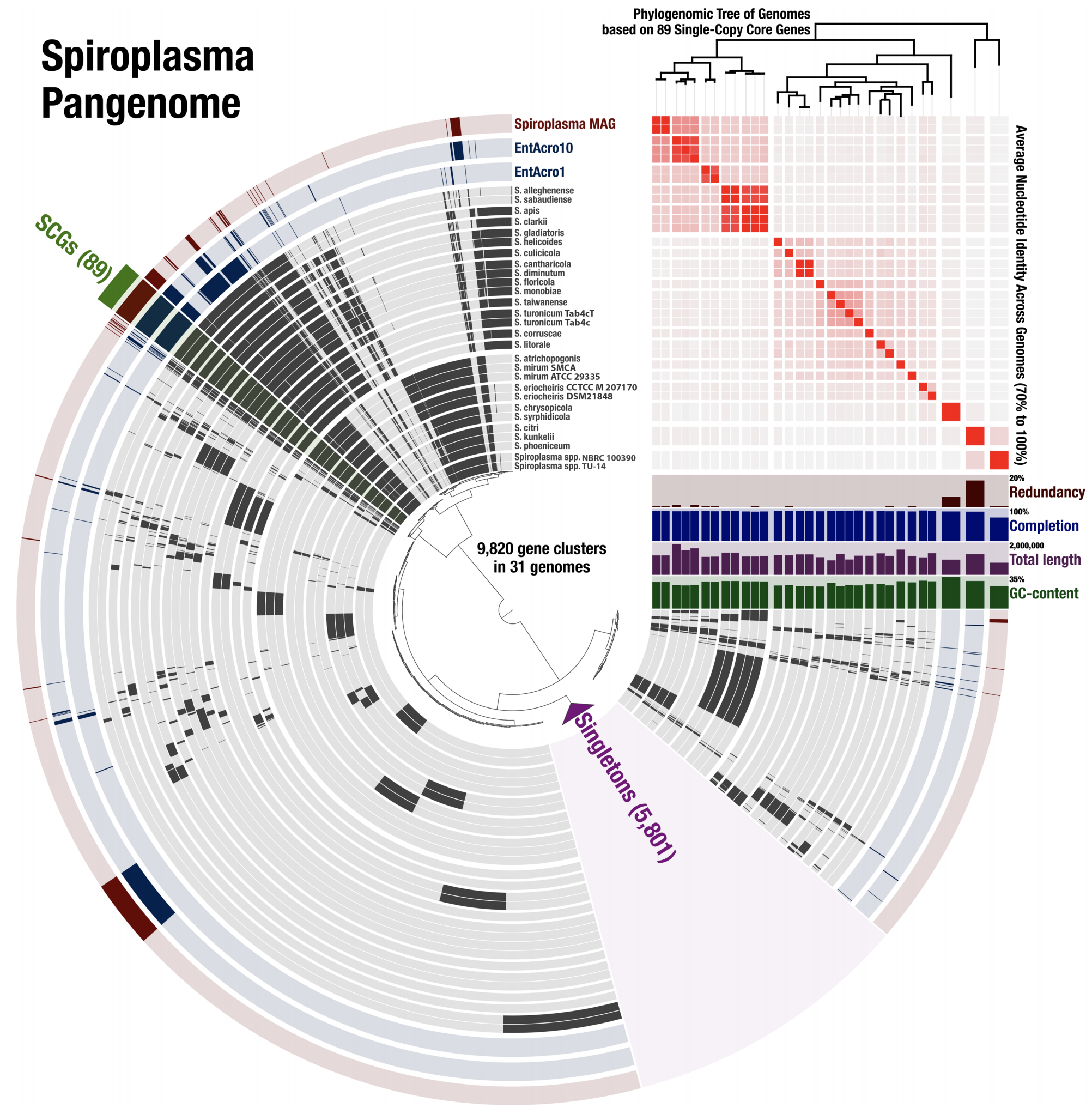 📚 PeerJ, 7(e7548) | 🔍 Google Scholar | 🔗 doi:10.7717/peerj.7548
📚 PeerJ, 7(e7548) | 🔍 Google Scholar | 🔗 doi:10.7717/peerj.7548
- A study that uses (1) genome-resolved metagenomics to reconstruct a population genome from fly metagenomes that resolves to the genus Spiroplasma, (2) pangenomics to put this genome in the context of other Spiroplasma genomes, (3) phylogenomics to infer ancestral relationships between Spiroplasma genomes, and (4) includes an ANI-based distance estimation between all genomes for comprehensive reporting.
- It is a particularly good example that demonstrates how pangenomics can reveal appropriate targets for high-resolution phylogenomics.
- A fully reproducible bioinformatics workflow for this multi'omics analysis is here. Anvi'o databases to interactively reproduce and explore the Spiroplasma pangenome is also available.
- It is a particularly good example that demonstrates how pangenomics can reveal appropriate targets for high-resolution phylogenomics.
- A fully reproducible bioinformatics workflow for this multi'omics analysis is here. Anvi'o databases to interactively reproduce and explore the Spiroplasma pangenome is also available.
Global phylogeography and ancient evolution of the widespread human gut virus crAssphage
 📚 Nature Microbiology, 4(10):1727 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-019-0494-6
📚 Nature Microbiology, 4(10):1727 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-019-0494-6
- A monumental effort lead by Rob Edwards and Bas Dutilh to explore with the help of 114 scientists from around the world the global phylogeorgraphy and evolution of crAssphage, one of the most numerous viruses in the human gut that infect bacteria.
- A curated list of press coverage of this study is available on Rob Edwards' web site.
- A curated list of press coverage of this study is available on Rob Edwards' web site.
Co-occurring genomic capacity for anaerobic methane and dissimilatory sulfur metabolisms discovered in the Korarchaeota
 📚 Nature Microbiology, 4(4):614-622 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-019-0362-4
📚 Nature Microbiology, 4(4):614-622 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-019-0362-4
- Yellowstone National Park, archaeal evolution, genome-resolved metagenomics, phylogenomics, and pangenomics (cool stuff all over).
- First description of a microbial population with both anaerobic methane and dissimilatory sulfur metabolisms.
- A news article by Montana State University with the photographs of cools scientists: 'An organism in Yellowstone hot spring potentially linked to earliest life on Earth'
- First description of a microbial population with both anaerobic methane and dissimilatory sulfur metabolisms.
- A news article by Montana State University with the photographs of cools scientists: 'An organism in Yellowstone hot spring potentially linked to earliest life on Earth'
Transcriptome-wide reprogramming of N 6-methyladenosine modification by the mouse microbiome
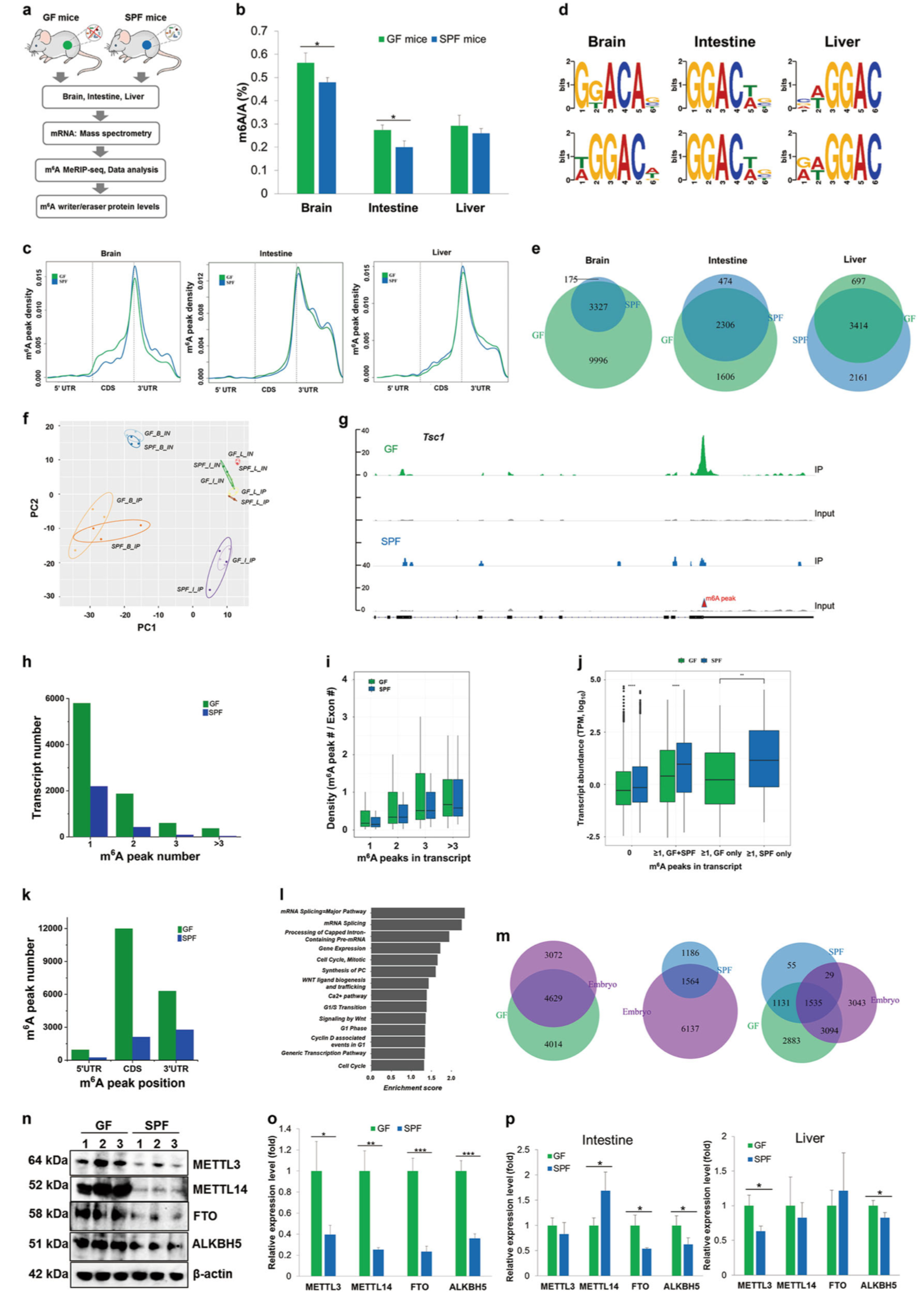 📚 Cell Research, 29:167–170 | 🔍 Google Scholar | 🔗 doi:10.1038/s41422-018-0127-2
📚 Cell Research, 29:167–170 | 🔍 Google Scholar | 🔗 doi:10.1038/s41422-018-0127-2
- N6-methyladenosine (m6A) is the most abundant mesenger RNA modification in mammalian cells, occurring at ~3 modified adenosine residues per transcript.
- Using liquid chromatography/mass spectrometry, this study shows differential occurrence of m6A modifications in brain, liver, and intestinal cells between germ-free and conventional mice, demonstrating that the microbiome has a strong effect on host m6A mRNA modification.
- Using liquid chromatography/mass spectrometry, this study shows differential occurrence of m6A modifications in brain, liver, and intestinal cells between germ-free and conventional mice, demonstrating that the microbiome has a strong effect on host m6A mRNA modification.
B cell superantigens in the human intestinal microbiota
📚 Science Translational Medicine, 11(507):eaau9356 | 🔍 Google Scholar | 🔗 doi:10.1126/scitranslmed.aau9356
2018
Microbiome characterization by high-throughput transfer RNA sequencing and modification analysis
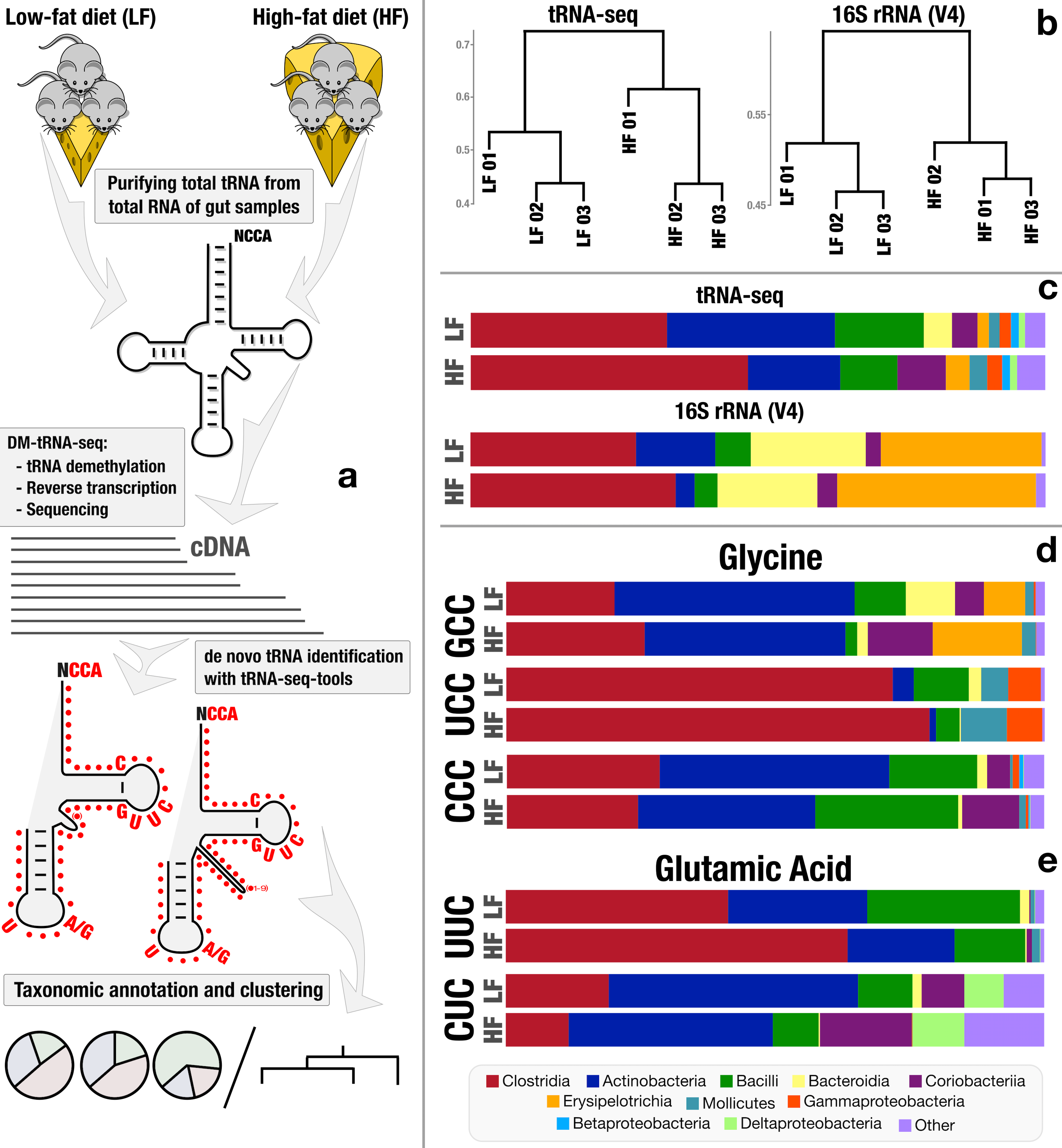 📚 Nature Communications, 9(1):5353 | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-018-07675-z
📚 Nature Communications, 9(1):5353 | 🔍 Google Scholar | 🔗 doi:10.1038/s41467-018-07675-z
- The first application of tRNA sequencing to environmental microbiomes (peer reviews and responses).
- Reveals taxon- and diet-dependent variations in tRNA modifications, and provides first in situ insights into 'metaepitranscriptomics' through tRNA gene expression dynamics and post-transcriptional modifications.
- New RNA sequencing strategy provides insight into microbiomes, by Matt Wood.
- RNA sequencing offers novel insights into the microbiome, by Liji Thomas, MD, and Kate Anderton, B.Sc.
- Reveals taxon- and diet-dependent variations in tRNA modifications, and provides first in situ insights into 'metaepitranscriptomics' through tRNA gene expression dynamics and post-transcriptional modifications.
- New RNA sequencing strategy provides insight into microbiomes, by Matt Wood.
- RNA sequencing offers novel insights into the microbiome, by Liji Thomas, MD, and Kate Anderton, B.Sc.
Nitrogen-fixing populations of Planctomycetes and Proteobacteria are abundant in surface ocean metagenomes
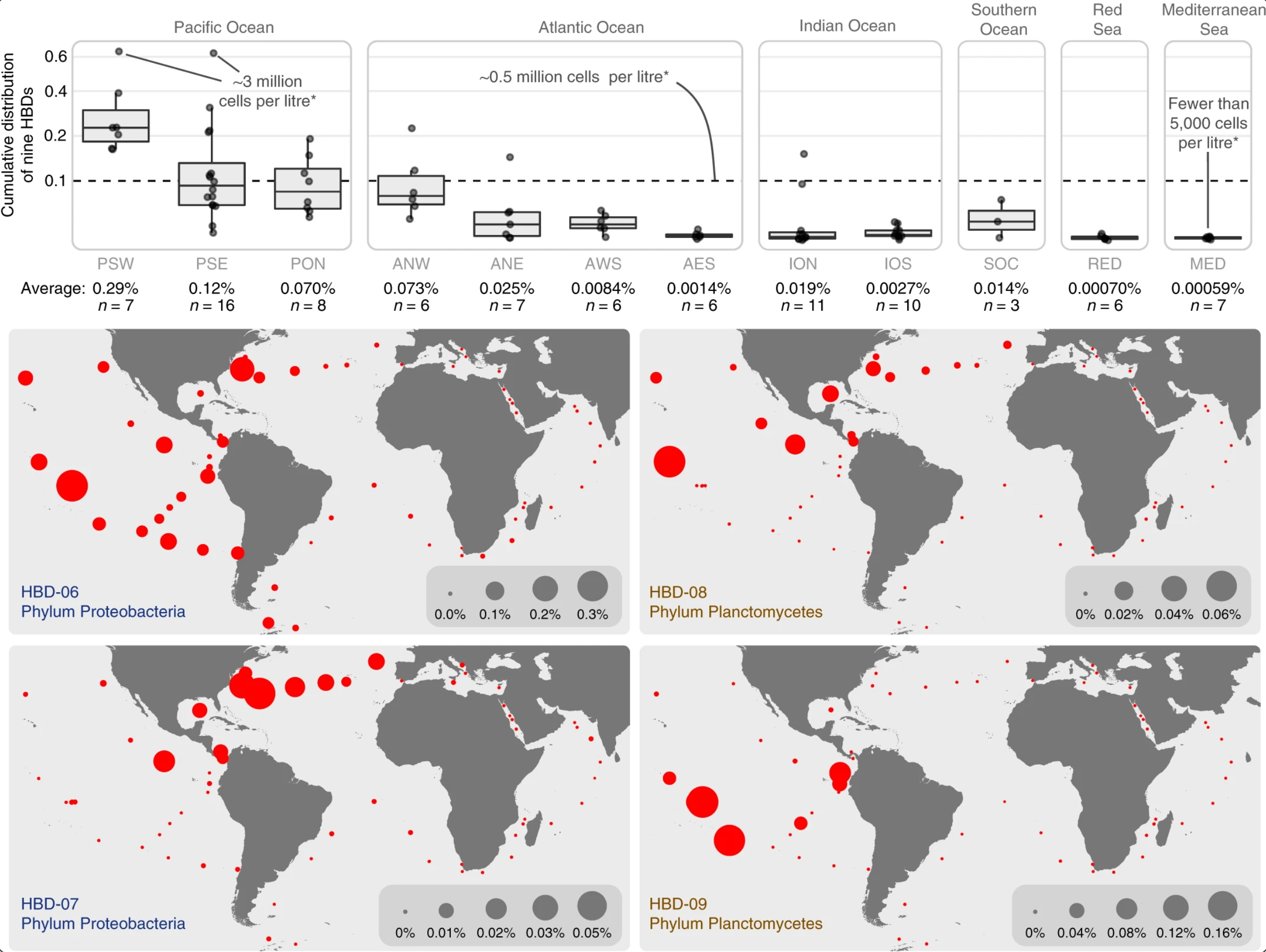 📚 Nature Microbiology, 3(7):804-813 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-018-0176-9
📚 Nature Microbiology, 3(7):804-813 | 🔍 Google Scholar | 🔗 doi:10.1038/s41564-018-0176-9
- First genomic evidence for abundant and widespread non-cyanobacterial nitrogen-fixing populations in the surface ocean.
- Nearly 1,000 non-redundant, high-quality bacterial, archaeal, and eukaryotic population genomes from TARA Oceans metagenomes.
- A "behind the paper" blog post by Meren, a press release by the MBL, and an extensive description of the bioinformatics workflow.
- Nearly 1,000 non-redundant, high-quality bacterial, archaeal, and eukaryotic population genomes from TARA Oceans metagenomes.
- A "behind the paper" blog post by Meren, a press release by the MBL, and an extensive description of the bioinformatics workflow.
Linking pangenomes and metagenomes -- the Prochlorococcus metapangenome
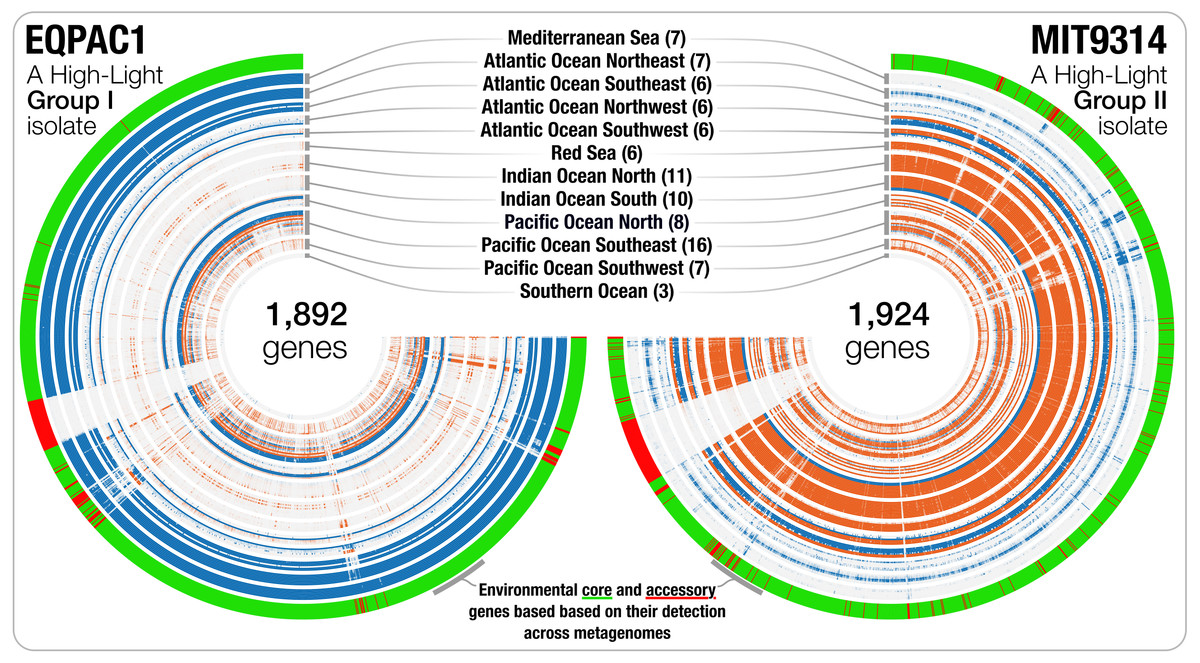 📚 PeerJ, 6(e4320) | 🔍 Google Scholar | 🔗 doi:10.7717/peerj.4320
📚 PeerJ, 6(e4320) | 🔍 Google Scholar | 🔗 doi:10.7717/peerj.4320
- A big-data study in which a pangenome of 31 Prochlorococcus isolates meets 31 billion Tara Oceans metagenomic sequences (Peer-review history).
- Metapangenomes reveal to what extent genes that may be linked to the ecology and fitness of microbes are conserved within a phylogenetic clade.
- Reproducible bioinformatics workflow.
- Metapangenomes reveal to what extent genes that may be linked to the ecology and fitness of microbes are conserved within a phylogenetic clade.
- Reproducible bioinformatics workflow.
Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host
📚 Nature, 557(7706):580–584 | 🔍 Google Scholar | 🔗 doi:10.1038/s41586-018-0125-z
- TET2 deficiency ➡️ deteriorating small intestinal barrier ➡️ bacterial translocation ➡️ incrased IL-6 signalling ➡️ pre-leukemic myeloproliferation (a leukemia precursor).
- ScienceDaily: Under certain conditions, bacterial signals set the stage for leukemia.
- ScienceDaily: Under certain conditions, bacterial signals set the stage for leukemia.
2017
Tracking microbial colonization in fecal microbiota transplantation experiments via genome-resolved metagenomics
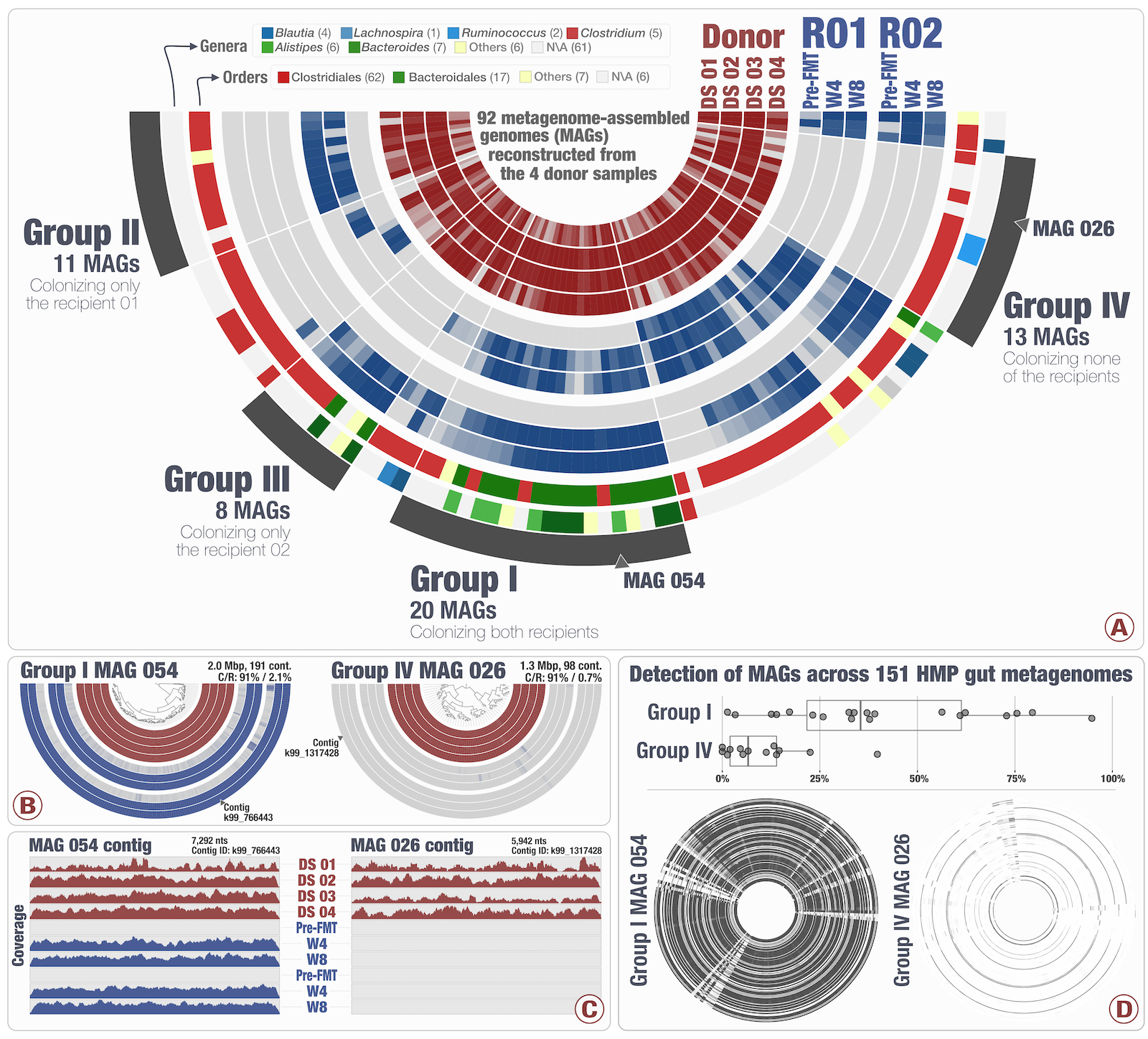 📚 Microbiome, 5(50) | 🔍 Google Scholar | 🔗 doi:10.1186/s40168-017-0270-x
📚 Microbiome, 5(50) | 🔍 Google Scholar | 🔗 doi:10.1186/s40168-017-0270-x
- An FMT study with metagenome-assembled genomes (see public data).
- Bacteroidales: high-colonization rate. Clostridiales: low colonization rate. Colonization success is negatively correlated with the number of genes related to sporulation.
- MAGs with the same taxonomy showed different colonization properties, highlighting the importance of high-resolution analyses.
- Populations colonized both recipients were also prevalent in the HMP cohort (and the ones that did not, distribute sporadically across the HMP cohort).
- Bacteroidales: high-colonization rate. Clostridiales: low colonization rate. Colonization success is negatively correlated with the number of genes related to sporulation.
- MAGs with the same taxonomy showed different colonization properties, highlighting the importance of high-resolution analyses.
- Populations colonized both recipients were also prevalent in the HMP cohort (and the ones that did not, distribute sporadically across the HMP cohort).
Simulations predict microbial responses in the environment? This environment disagrees retrospectively
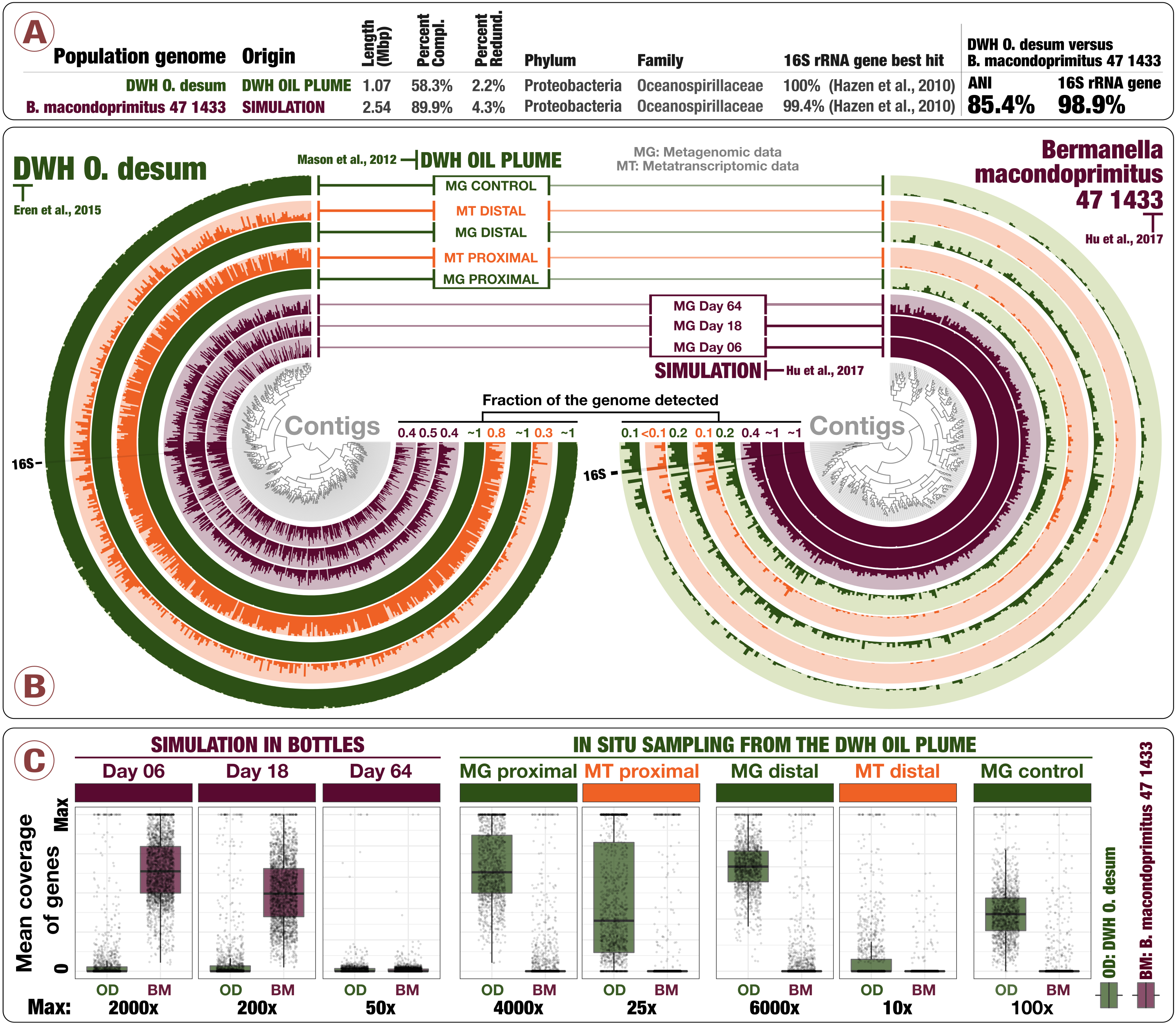 📚 Proceedings of the National Academy of Sciences, 114(43):E8947-E8949 | 🔍 Google Scholar | 🔗 doi:10.1073/pnas.1712186114
📚 Proceedings of the National Academy of Sciences, 114(43):E8947-E8949 | 🔍 Google Scholar | 🔗 doi:10.1073/pnas.1712186114
- A letter that re-analyzes some of the findings published in Hu et al.
- Here is a rebuttal from Probst et al. challenging our findings in this letter.
- Here is our response to Probst et al., and the recovery of DWH O. Desum v2.
- Here is a rebuttal from Probst et al. challenging our findings in this letter.
- Here is our response to Probst et al., and the recovery of DWH O. Desum v2.
DESMAN: a new tool for de novo extraction of strains from metagenomes
📚 Genome Biology, 18(1):181 | 🔍 Google Scholar | 🔗 doi:10.1186/s13059-017-1309-9
Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea
📚 Nature biotechnology, 35(8):725 | 🔍 Google Scholar | 🔗 doi:10.1038/nbt.3893
Peripartum antibiotics promote gut dysbiosis, loss of immune tolerance, and inflammatory bowel disease in genetically prone offspring
 📚 Cell Reports, 20(2):491-504 | 🔍 Google Scholar | 🔗 doi:10.1016/j.celrep.2017.06.060
📚 Cell Reports, 20(2):491-504 | 🔍 Google Scholar | 🔗 doi:10.1016/j.celrep.2017.06.060
- Antibiotics during pregnancy promote offspring gut dysbiosis, immune dysfunction, and IBD.
- Antibiotics given after the developmental period do not increase IBD.
- Antibiotic-perturbed maternal microbiota likely contribute to neonatal gut dysbiosis.
- Press release.
- Antibiotics given after the developmental period do not increase IBD.
- Antibiotic-perturbed maternal microbiota likely contribute to neonatal gut dysbiosis.
- Press release.
2016
Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies
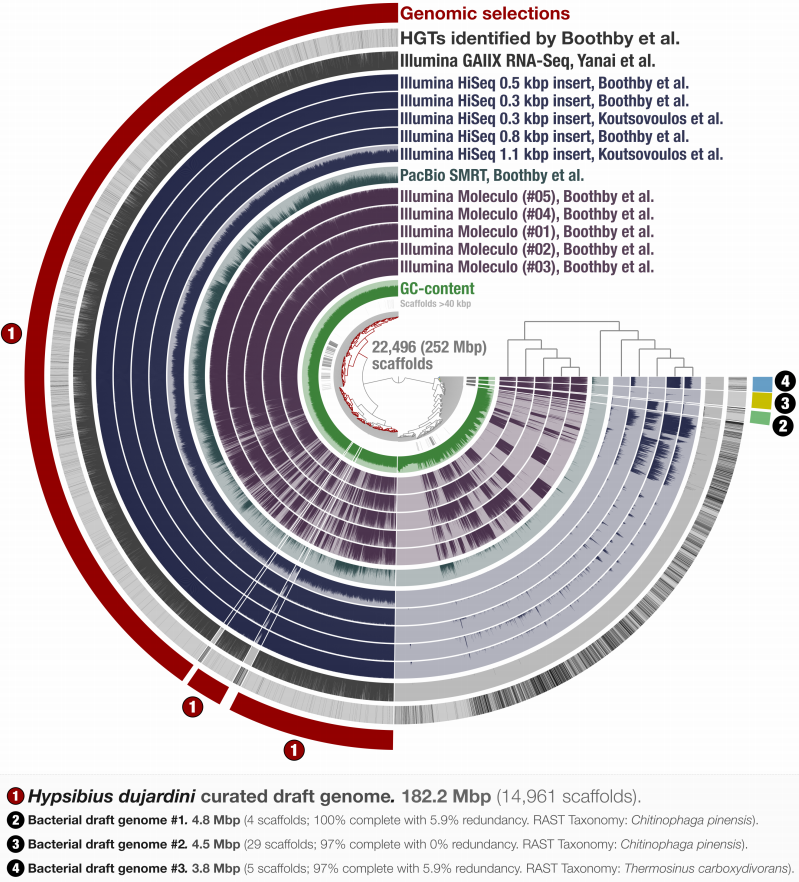 📚 PeerJ, 4:e1839 | 🔍 Google Scholar | 🔗 doi:10.7717/peerj.1839
📚 PeerJ, 4:e1839 | 🔍 Google Scholar | 🔗 doi:10.7717/peerj.1839
- A holistic approach to visualize and curate genomic and metagenomic assemblies.
- A re-analysis of the first released Tardigrade genome reveals a likely symbiont among other contaminants.
- A practical approach to estimate the number bacterial genomes in an assembly.
- A re-analysis of the first released Tardigrade genome reveals a likely symbiont among other contaminants.
- A practical approach to estimate the number bacterial genomes in an assembly.
New insights into microbial ecology through subtle nucleotide variation
📚 Frontiers in microbiology, 7:1318 | 🔍 Google Scholar | 🔗 doi:10.3389/fmicb.2016.01318
2015
Anvi'o: an advanced analysis and visualization platform for ‘omics data
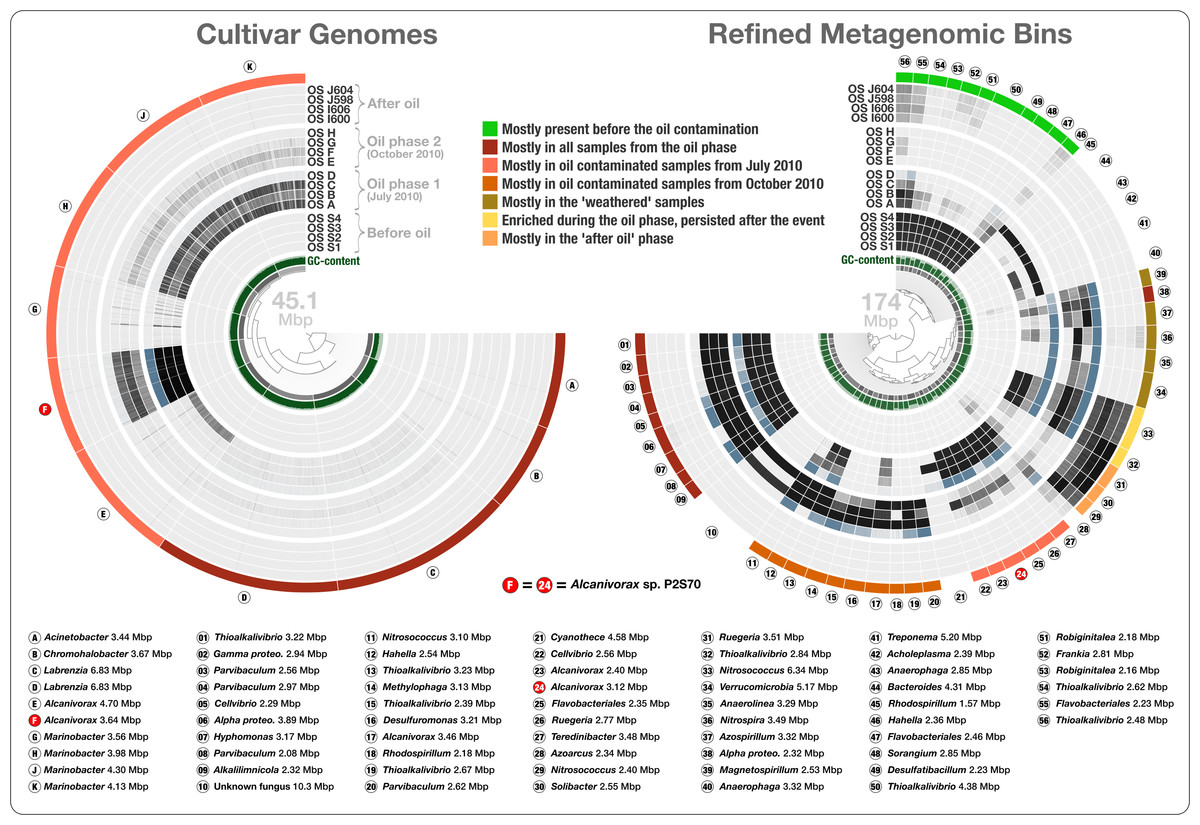 📚 PeerJ, 3:e1319 | 🔍 Google Scholar | 🔗 doi:10.7717/peerj.1319
📚 PeerJ, 3:e1319 | 🔍 Google Scholar | 🔗 doi:10.7717/peerj.1319
- The very first description of anvi'o.
- Demonstrating its abilities in genome-resolved metagenomics, and microbial population genetics via single-nucleotide variant analyses across metagenomes.
- Re-analysis of cultivar genomes, metagenomes, and metatranscriptomes associated with the Deepwater Horizon oil spill.
- Demonstrating its abilities in genome-resolved metagenomics, and microbial population genetics via single-nucleotide variant analyses across metagenomes.
- Re-analysis of cultivar genomes, metagenomes, and metatranscriptomes associated with the Deepwater Horizon oil spill.
Minimum entropy decomposition: unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences
📚 The ISME Journal, 9(4):968 | 🔍 Google Scholar | 🔗 doi:10.1038/ismej.2014.195
A single genus in the gut microbiome reflects host preference and specificity
📚 The ISME Journal, 9(1):90 | 🔍 Google Scholar | 🔗 doi:10.1038/ismej.2014.97
2014
Oligotyping analysis of the human oral microbiome
📚 Proceedings of the National Academy of Sciences, 111(28):E2875-E2884 | 🔍 Google Scholar | 🔗 doi:10.1073/pnas.1409644111
2013
Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data
📚 Methods in Ecology and Evolution, 4(12):1111-1119 | 🔍 Google Scholar | 🔗 doi:10.1111/2041-210X.12114