Chapter I - Reproducing Kiefl et al, 2022

Table of Contents
Quick Navigation
- Chapter I: The prologue ← you are here
- Chapter II: Configure your system
- Chapter III: Build the data
- Chapter IV: Analyze the data
- Chapter V: Reproduce every number
Introduction
This multi-part document details the story of how our paper came to be. As a reminder, you can always find the published version of the paper here:
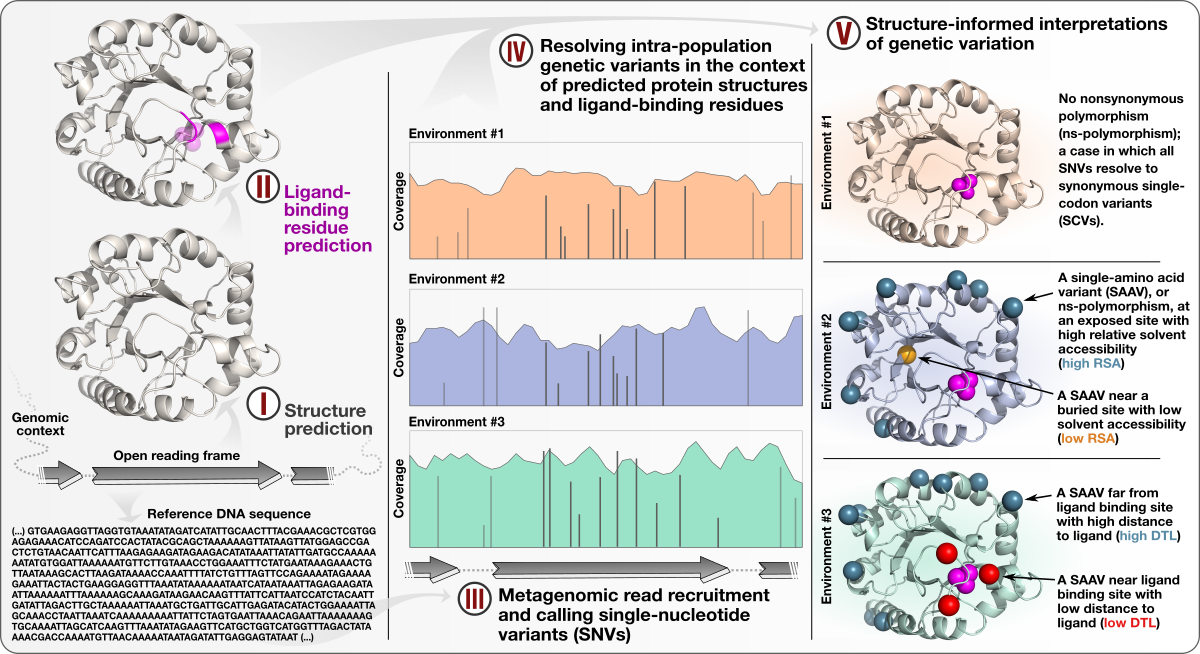
- Demonstrates a quantifiable link between (1) the magnitude of selective pressures over key metabolic genes (e.g., glutamine synthase of the central nitrogen metabolism), (2) the availability of key nutrients in the environment (e.g., nitrate), and (3) the maintenance of nonsynonymous variants near protein active sites.
- Shows that the interplay between selective pressures and protein structures also maintains synonymous variants -- revealing a quantifiable link between translational accuracy and fluctuating selective pressures.
- Comes with a reproducible bioinformatics workflow that offers detailed access to computational steps used in the study that spans from metagenomic read recruitment and profiling to the integration of environmental variants and predicted protein structures.
This document includes step-by-step procedures for reproducing our study, in-depth explanations of our analysis decisions, and technical details that were not suitable for the main text or supplemental info.
Philosophy
A central tenant of the scientific process is reproducibility, yet we are in the midst of a reproducibility crisis in which scientists constantly fail to reproduce published results.
Despite the computer being governed by deterministic rules, computational sciences are not immune to issues of irreproducibility. Only with great care and foresight can a computationally-driven study be considered reproducible. And unfortunately, it is rare to find studies that provide reproducible documents comprehensive enough to replicate even the central findings of the study. Of course this isn’t due to ill intent (in the assumed vast majority of cases), but instead due to a lack of time to create such a document, and/or a lack of skills required to make it right.
Indeed, writing this document took a ridiculous amount of time, and structuring my data analysis with reproducibility in mind took several iterations of planning and execution. I have done this analysis not once, but countless times–on different machines and operating systems. And despite all of my efforts, there is tons of room for improvement. This is because the reproducible ceiling is so high. Let’s talk more about that.
To some, the holy grail of reproducible workflows is a program script that I’ll call generate_paper.sh, that when run, downloads all raw data, runs all analyses, creates all figures, tables and numbers, plops them into a LaTeX file where the paper’s text is sitting, and compiles a pdf of the paper.
What value does this bring? Well, if a reviewer wants a statistical analysis different from the one used, just open up generate_paper.sh, find the relevant section, change pearson to spearman, and regenerate the paper by typing ./generate_paper.sh. Or if a reader is intrigued by the implications of a result, they can run ./generate_paper.sh, find the intermediate files, and extend the analysis to their liking.
The value of generate_paper.sh is therefore understandably enormous. But there’s a missing element to this, isn’t there? It’s the human element, the part that explains why decisions are being made, as opposed to just a detailed implementation. This is equally important as reproducibility. And unfortunately, manuscript conventions for conciseness oftentimes push nitty gritty details out of the paper. With no regulated medium for these details to be archived, they more often than not end up cast into the wind and ultimately lost.
With this in mind, this document aims to explain both the how and the why of our research, by intermingling step-by-step reproducibility with in-depth commentary. This is because in our experience readers are much more interested in drawing inspiration for their own research than they in replicating our results, and this document is structured to reflect this.
Goals
As stated, this document is a co-mingling of step-by-step reproducibility, and in-depth commentary. With that in mind, I set forth the following goals, which in my opinion are all satisfied by this document:
- Step-by-step command-line instructions are provided for generating all raw, intermediate, and final data used in the paper, including all figures, tables, and numbers in the main and supplemental text.nal
- There exists a ‘data checkpoint’, so that one may download intermediate data if they can’t or don’t want to follow the entirety of the workflow. This is especially important for steps requiring significant computational resources. For example, this workflow involves downloading roughly 4Tb of metagenomic and metatranscriptomic sequence data for a read recruitment experiment. Yet those unable or uninterested in allotting this amount of storage space can instead download the anvi’o databases (a few Gb) that result from this read recruitment experiment and continue with the more interesting aspects of the analysis.
- I’ve avoided common pitfalls of replicability by paying extra attention to (a) creating isolated installation environments, (b) meticulous control over program versioning, (c) cross-platform compatibility, and (d) prohibited use of environmental variables that are specific to my personal environment.
- Let’s face it: there isn’t enough space in the Methods section for all relevant details. Beyond what’s required for reproducibility, this document houses information about methods that didn’t make the cut, but are important nonetheless.
- In my opinion, this document sets a standard for what transparency in computational science means, and why that’s so valuable. This analysis is completely laid bare, so that it may be openly challenged, criticized, riffed on, and hopefully improved upon in the process. It likely contains errors–but that’s okay. I didn’t make this document to prove everything I did was right. I made this document to be educational.
Requirements
There are some skill and hardware requirements for following this workflow. There are also software requirements, however after completing the first step in this workflow, it is guaranteed that all of your software requirements are met.
Hardware
This study makes primary use of some large datasets. If you want to reproduce this study from scratch, you will need access to a high performance computing (HPC) cluster.
But if you don’t have access to such a resource, don’t worry. All of the computationally intensive steps in this study occur near the beginning of the workflow. After Step 4, I achieved all of the subsequent analyses on a laptop with 16 Gb of RAM.
Skillset
This workflow will use language that assumes you have basic knowledge of UNIX (command-line) based scientific computing.
That means you know the basics of navigating to different files on your computer using commands like cd and ls. It means you shouldn’t be surprised that if you have an R script called test.R, you could execute that script with the following syntax: Rscript test.R.
If you don’t fall into this category of people, I would suggest checking out this great learning resource, Happy Belly Bioinformatics, which teaches you the basics of UNIX command-line computing.
If you are concerned whether your skills are up to snuff, don’t worry. My suggestion would be to briefly peruse Chapter II: Configure your system. This is by far the hardest part of the workflow, so if you think you can follow the steps within, the rest will be child’s play.
As you learned in the hardware requirements, recreating this study from scratch requires access to a high-performance computing cluster. If you plan to go this route instead of downloading the checkpoint datapack, you will need to know how to submit commands as jobs to your HPC cluster.
Format
I tried to adopt a consistent terminology throughout the document, so please take the time to learn it below.
Workflow anatomy
The meat of the workflow is composed of steps and analyses.
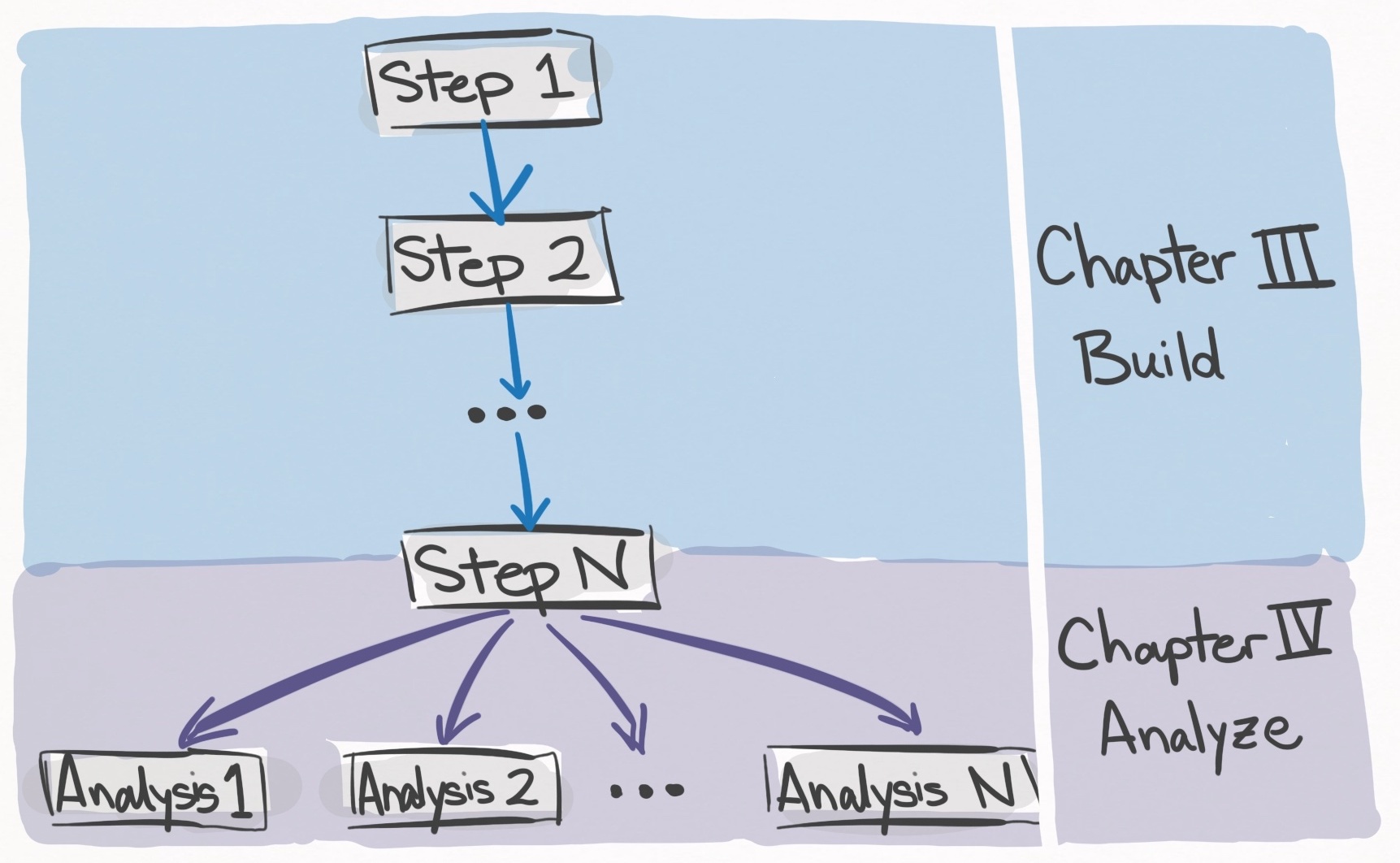
In the simplest terms, steps build up the data, and analyses utilize the data.
Chapter III: Build the data (and Chapter II: Configure your system) is composed of a series of steps which are to be completed in order. Each step tackles a discrete task such as “downloading the metagenomes” or “annotating genes with functions”. Once the last step has been completed, you will possess all of the necessary data structures to carry out the analyses in the paper.
In contrast, Chapter IV: Analyze the Data is composed of analyses which can be completed independently of one another (unless otherwise stated). Each analysis represents a discrete analysis like “calculating codon usage between SAR11 genes” or “comparing AlphaFold to MODELLER”. Each analysis uses the data structures built from Chapter II, and thus all of the steps must be completed before analysis can be completed. Now let’s talk more about the formatting inside each step and analysis.
Command
Each step and analysis is composed of a series of commands. Ideally, you should be able to run these commands in succession by copy-pasting them into your command-line.
To distinguish these commands from all other commmentary and code, I have put each command inside a red box. In addition to the command itself, each red box contains the time and storage requirements for running the command. If the command requires internet, I add the flag: ‘‣ Internet: Yes’. If the command should be submitted as a job due to high memory/storage/time requirements, I add the flag: ‘‣ Cluster: Yes’. Here is an example command:
Command #X
anvi-split -C GENOMES \
-c 03_CONTIGS/SAR11_clade-contigs.db \
-p 06_MERGED/SAR11_clade/PROFILE.db \
-o 07_SPLIT
‣ Time: 90 min
‣ Storage: 28.3 Gb
If there are sections of commands that need to be edited manually by you, required edits will be denoted within angular brackets (<>). For example,
Command #Y
cd <A_DIRECTORY_YOU_LIKE>
mkdir kiefl_et_al_2021
‣ Time: <1 min
‣ Storage: Minimal
In this case, <A_DIRECTORY_YOU_LIKE> refers to a directory you decide. For example, I might replace it with /Users/evan/Desktop.
Help is a click away
Purple keywords refer to anvi’o concepts that have help pages. For example, contigs-db refers to an anvi’o contigs database that can be clicked on to learn more about.
Green keywords refer to anvi’o programs that have help pages. For example, anvi-gen-contigs-database refers to an anvi’o program that can create an anvi’o contigs database.
The links within these documents lead to other anvi’o concepts, which lead to other anvi’o concepts. It’s kind of like Wikipedia–you can get lost in it. Because many concepts already have rich documentation, I often forego in-depth explanations, assuming you will click the link to learn more.
Directory
With that all out of the way, it is time for the workflow itself. Due to its size, I’ve broken it up into several parts, each one being a separate document. At the top of document you can find a directory for quickly switching between all of the different parts. Below is a description of each document.
Chapter I: The prologue
This is what you’re reading right now. In this section the philosophy, goals, expectations, requirements, and gameplan are all laid out.
Chapter II: Configure your system
In this section, you’ll set up your computational environment very precisely so that everything downstream runs as expected and you don’t run into errors. This is the hardest chapter of the the workflow because exactness is key. But you’ll be walked through each step, so don’t worry.
Chapter III: Build the data
In this section, you’ll start with nothing but the internet, and build up all of the required data structures needed to reproduce all of the analyses in the paper.
Chapter IV: Analyze the data
In this section, you’ll utilize all of the data structures you’ve worked so hard to build in Chapter III, and reproduce each analysis in the study (or just the one’s you care about).
Chapter V: Reproduce every number
In this section, you’ll be provided the required scripts to reproduce every mentioned number in the main text and supplemental text of the study.