


Phylogenomics of SAR11

Table of Contents
The purpose of this document, which is written by Meren together with Kelle Freel, is to provide its readers with a reproducible workflow that can be used to regenerate or extend the SAR11 phylogenomic tree that appeared in the manuscript below:
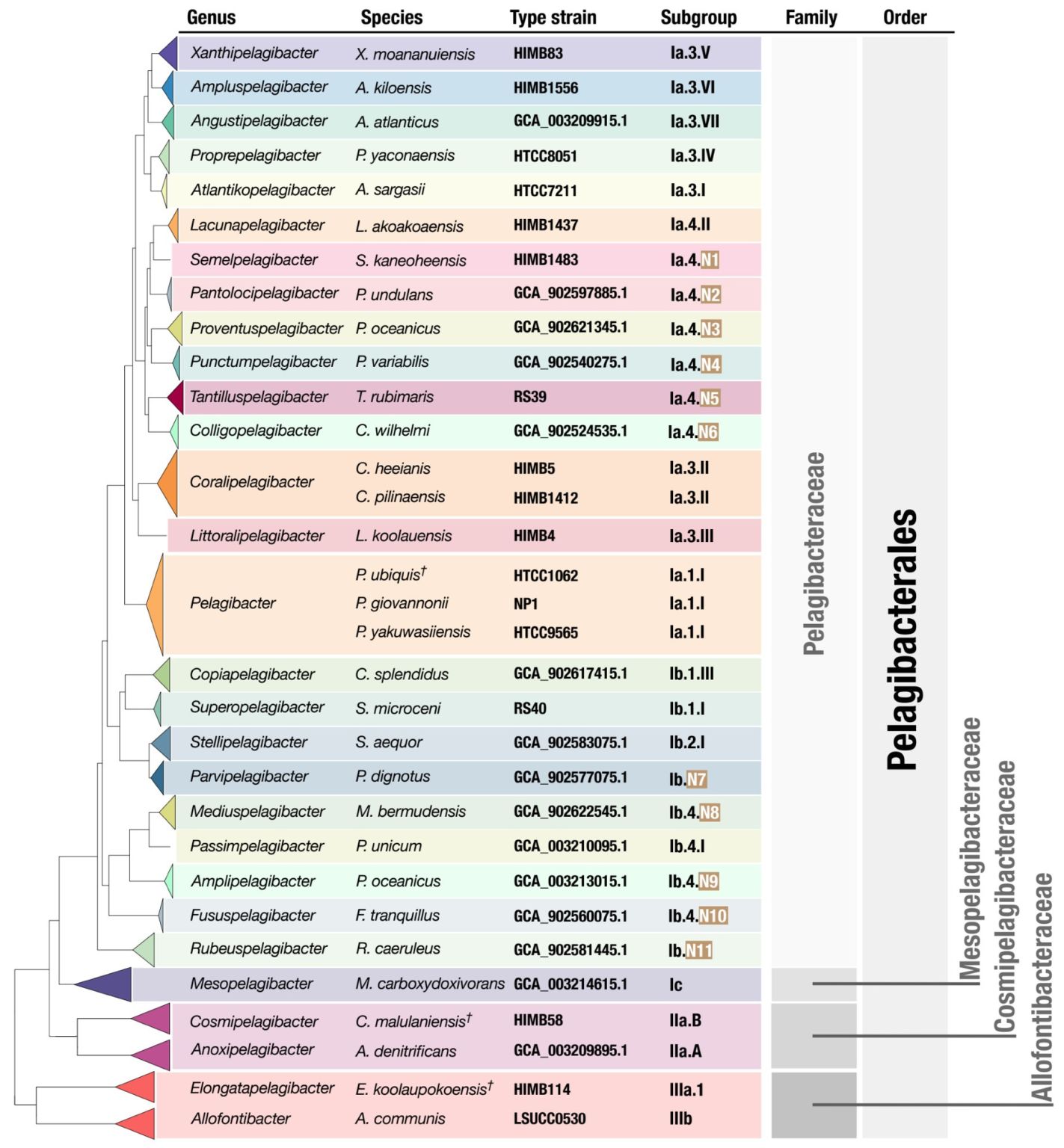
- Uses the new high-quality SAR11 genomes to propose a taxonomic framework for SAR11 by introducing 24 genera within the family Pelagibacteraceae along with formal names for each one of them, bringing a much-needed order to SAR11 taxonomy and opens the door for community-wide discussions to resolve the nomenclature of this clade.
- You can find a little write-up by Meren on LinkedIn here.
For your technical questions about the tutorial, please feel free to reach out to Meren or Kelle. Please reach out to Michael Rappé for things that may require input from the senior author of our work.
Data. Now!
For those of you who are in a rush, and more interested in the data items than the bioinformatics, here is the list:
- doi:10.6084/m9.figshare.30464900: All FASTA files for each 494 SAR11 genomes.
- doi:10.6084/m9.figshare.30467441: All anvi’o contigs-db files for each 494 SAR11 genomes (annotated with all sorts of HMMs and gene functions, ready for downstream analyses).
- genome-data.txt.gz: Genome metadata.
- Alphaproteobacterial_SCGs.tar.gz: HMMs for alphaproteobacterial single-copy core genes (SCGs) in anvi’o hmm-source format (in which genes.hmm.gz contains each model).
- PRJNA1170004: NCBI SRA accession for the whole genome shotgun sequencing of cultures for new isolate genomes.
Thank you for your interest.
A brief introduction to Freel et al.
Members of SAR11, or the Pelagibacterales, dominate the global surface ocean and is key to the marine carbon cycle. To understand the specific roles that this incredibly diverse clade of marine bacteria has, it is crucial to have high quality isolate genomes. In Freel et al., we focused on isolating hundreds of SAR11 from the coastal and offshore surface seawater of the tropical Pacific Ocean and generated 81 high-quality genomes. Exploring these genomes and their global distributions using metagenomic data, allowed us to define discrete Pelagibacterales ecotypes that we defined as genera.
In order to unify SAR11 nomenclature, we have provided names for the genera we identified, while this does not exhaustively account for all of the identified SAR11 diversity, we think that this is a step towards a meaningful hierarchical system for this important marine bacteria. Integrating evolution and ecology, into this hierarchical system is essential to better understand the enigmatic and incredibly abundant Pelagibacterales.
The goal of this documentation is to provide an accessible method to reproduce the SAR11 phylogeny generated in our publication titled “New isolate genomes and global marine metagenomes resolve ecologically relevant units of SAR11” in order to place additional high-quality SAR11 genomes in the structure we have established.
How to follow this tutorial
This tutorial will take you step by step from downloading the raw data for genomes in our study in FASTA format all the way down to the tree calculations and visualizations.
You can copy-paste all the commands to your terminal, but please read everything carefully.
It would be best if you follow the tutorial using an anvi’o-installed computer (whether it is your laptop or a server). For the sake of convenience, this is how you can start:
# I will setup everyting in my home directory, but you can replace `$HOME` below
# with any directory full-path to get things going
export workdir=$HOME/sar11-phylogenomics
# create the directory if it doesn't exist
mkdir -p $workdir
# go into it to get things going
cd $workdir
If you are here, when you type ls in your terminal and press ENTER, you shouldn’t see an output (so that you are in a new, empty directory).
And similarly, when you run anvi-self-test the following way, you should be getting an output similar to this:
anvi-self-test -v
Anvi'o .......................................: marie (v8-dev)
Python .......................................: 3.10.15
Profile database .............................: 40
Contigs database .............................: 24
Pan database .................................: 21
Genome data storage ..........................: 7
Structure database ...........................: 2
Metabolic modules database ...................: 4
tRNA-seq database ............................: 2
Genes database ...............................: 6
Auxiliary data storage .......................: 2
Workflow configurations ......................: 4
If that is the case, you are golden.
You can follow this tutorial in two ways:
- Downloading all the FASTA files for the SAR11 genomes used in Freel et al, and do everything from scratch (which will take more time, but you will feel like a hacker).
- Downloading all the anvi’o contigs-db files generated from these genomes, and do everything after that (which will be more convenient, but you will feel like, well, happy, I guess).
In both ways, you will have ways to extend the collection of genomes with your own, either with FASTA files or contigs-db files you may already have for your genomes.
Downloading FASTA files
We used 494 genomes in Freel et al, and you can download them from doi:10.6084/m9.figshare.30464900 and unpack them by running the following commands:
# download the FASTA files
curl -L -o GENOMES.tar.gz \
-H "User-Agent: Chrome/115.0.0.0" \
https://figshare.com/ndownloader/files/59115383
# unpack it
tar -zxvf GENOMES.tar.gz
# get rid of the downloaded file
rm -rf GENOMES.tar.gz
# make sure everything looks ok, and the output of the
# following command is 494
ls GENOMES/*fa | wc -l
If you are here, it means you have all 494 FASTA files on your computer. Of these 494,
- 375 are single-amplified genomes (SAGs),
- 81 are new isolate genomes (FASTA) by Freel et al.,
- 28 are isolate genomes from previous efforts,
- and 10 are here to serve as outgroups.
Please run the following command to download this metadata table:
# download the file to our work directory
curl https://merenlab.org/data/sar11-phylogenomics/files/genome-data.txt.gz -o genome-data.txt.gz
# uncpack it
gzip -d genome-data.txt.gz
Now if you take a look at the genome-data.txt file, you will see that it shows a bunch of information for the genomes you have downloaded:
genome |
mapping_rep_accession |
subclade |
genus_species |
accession |
genome_extended |
genome_type |
is_95ANI_rep |
isolate_origin |
|---|---|---|---|---|---|---|---|---|
| HTCC1040 | HTCC1062 | Ia.1.I | Pelagibacter | HTCC1040 | HTCC1040 | isolate | NA | other |
| HTCC1062 | HTCC1062 | Ia.1.I | Pelagibacter_ubiqueversans | HTCC1062 | HTCC1062 | isolate | TRUE | other |
| HTCC9565 | HTCC9565 | Ia.1.I | Pelagibacter_yakuwasiensis | HTCC9565 | HTCC9565 | isolate | TRUE | other |
| NP1 | NP1 | Ia.1.I | Pelagibacter_giovannonii | NP1 | NP1 | isolate | TRUE | other |
| HTCC7211 | HTCC7211 | Ia.3.I | Atlantikopelagibacter_sargassoseensis | HTCC7211 | HTCC7211 | isolate | TRUE | other |
| HTCC7214 | HTCC7214 | Ia.3.I | Atlantikopelagibacter | HTCC7214 | HTCC7214 | isolate | TRUE | other |
| (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) |
| LSUCC0723 | LSUCC0723 | IIIa | III | 2739368061 | LSUCC0723 | isolate | TRUE | other |
| LSUCC0664 | LSUCC0664 | IIIa | III | 2770939455 | LSUCC0664 | isolate | TRUE | other |
| (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) |
Here are what each of these columns mean:
genome: The name of the genome. Should matchGENOMES/NAME.faorCONTIGS-DBs/NAME.dbin later stages of this tutorial.mapping_rep_accession: indicating whether the genome is the 95% ANI representative for the clade it belongs.subclade: which SAR11 subclade of the historical nomenclature best explains the genome.genus_species: the genus designation, if defined in Freel et al., and the species name, if one assigned for this particular genome.accession: accession number for the genome. This is a bit finicky. It can be ENA/NCBI accession, or IMG accession for a few that were not available in public resources.genome_extended: if the accession included additional information, we kept that information in this column.genome_type: whether the genome is an isolate or a SAG.is_95ANI_rep: whether the genome was a representative for the read recruitment analyses in Freel et al.isolate_origin: whether the isolate is from Freel et al
If you wish to extend the phylogenomic analysis with your own FASTA files, this is the time to add each genome you have into the GENOMES directory, and extend the contents of this file with entries like this:
genome |
mapping_rep_accession |
subclade |
genus_species |
accession |
genome_extended |
genome_type |
is_95ANI_rep |
isolate_origin |
|---|---|---|---|---|---|---|---|---|
| MY_GENOME_NAME_01 | UNKNOWN | NEW_GENOME | ||||||
| MY_GENOME_NAME_02 | UNKNOWN | NEW_GENOME |
Please use simple names for your entries to extend this table (without spaces and weird characters except _), and make sure there is a FASTA file with the same name and .fa extension in the GENOMES directory. If you follow these, everything below work for you smoothly, and the final tree will include your genomes, and by searching for ‘UNKNOWN’ subclades in the final visualization, you will be able to determine the best matching name/clade information for your genomes.
Getting the contigs-db files
Analyses described here require us to generate an anvi’o contigs-db for each FASTA file in the GENOMES directory.
You can either,
- OPTION #1: Generate contigs-db files from scratch, which is the appropriate way to do it if you added new FASTA files to the
GENOMESdirectory, or, - OPTION #2: Download all the previously generated contigs-db files, which will give you access to the same files if you were to go with the first option.
OPTION 1: Generate contigs-dbs from scratch
So you want to generate your own contigs-db files from scratch. Good for you. Now, there are two ways to do it (this tutorial is basically a labyrinth like that).
The first option is to do it on your own computer like a farmer from 18th century. Here is a for loop in BASH that will make that happen for you using the program anvi-gen-contigs-database to turn your fasta into a contigs-db and then annotate it with a few anvi’o programs such as anvi-run-hmms, anvi-run-ncbi-cogs, anvi-run-kegg-kofams, and anvi-run-scg-taxonomy:
# generate a directory for CONTIGS-DBs
mkdir -p CONTIGS-DBs
num_threads=8
for fasta in GENOMES/*.fa
do
# let's get the genome name
genome=$(basename "$fasta" .fa)
# generate a contigs-db
anvi-gen-contigs-database -f GENOMES/$genome.fa -o CONTIGS-DBs/$genome.db
# annotate the contigs-db file with some useful information
anvi-run-hmms -c CONTIGS-DBs/$genome.db --num-threads $num_threads
anvi-run-ncbi-cogs -c CONTIGS-DBs/$genome.db --num-threads $num_threads
anvi-run-kegg-kofams -c CONTIGS-DBs/$genome.db --num-threads $num_threads
anvi-run-scg-taxonomy -c CONTIGS-DBs/$genome.db --num-threads $num_threads
done
But of course this will take forever (more precisely, about 4 minutes per genome on a laptop, and about 33 hours for all). That’s why you actually would like to to do it on your HPC like a proper modern human indivdiual. But every HPC is different, so it is difficult for me to give you copy-pasta instructions here. On our own HPC, which is using slurm, I would have created a script in my work directory with the following content, and have named it gen-contigs-db.sh,
num_threads=8
# get the genome name from the user parameter
genome=$(basename $1 .fa)
# generate a contigs-db
anvi-gen-contigs-database -f GENOMES/$genome.fa -o CONTIGS-DBs/$genome.db
# annotate the contigs-db file with some useful information
anvi-run-hmms -c CONTIGS-DBs/$genome.db --num-threads $num_threads
anvi-run-ncbi-cogs -c CONTIGS-DBs/$genome.db --num-threads $num_threads
anvi-run-kegg-kofams -c CONTIGS-DBs/$genome.db --num-threads $num_threads
anvi-run-scg-taxonomy -c CONTIGS-DBs/$genome.db --num-threads $num_threads
and then I would have sent all the jobs to our cluster using the conveninence of our slurm wrapper, clusterize, with the following for loop:
mkdir -p CONTIGS-DBs
for fasta in GENOMES/*fa
do
clusterize "bash gen-contigs-db.sh $fasta" -n 8
done
Which would generate all the contigs-db files in the CONTIGS-DBs directory (in about 4 minutes).
I hope it is clear to you how to run this on your own HPC using this information, but if not, you have three options:
- Take my Programming for Life Scientsts course next year to master all these things,
- Contact your local HPC help as they should be able to easily figure it out what to do from the information here,
- Reach out to me directly and ask for help if you are really desperate, but I hope it will not come to this as it is very difficult to solve HPC releted issues remotely.
OPTION 2: Download pre-generated contigs-db files
Well, if you are here, it means you just want to get all the contigs-db files that we have generated following the workflow described above.
Luckily you can download it from doi:10.6084/m9.figshare.30467441, and unpack that directory using the following command:
# download the contigs-db files
curl -L -o CONTIGS-DBs.tar.gz \
-H "User-Agent: Chrome/115.0.0.0" \
https://figshare.com/ndownloader/files/59126048
# unpack it
tar -zxvf CONTIGS-DBs.tar.gz
# get rid of the downloaded file
rm -rf CONTIGS-DBs.tar.gz
# make sure everything looks ok, and the output of the
# following command is 494
ls CONTIGS-DBs/*db | wc -l
If you are here, running the anvi-db-info on any contigs-db in your work directory,
anvi-db-info CONTIGS-DBs/FZCC0015.db
should yield and output that looks like this:
DB Info (no touch)
===============================================
Database Path ................................: CONTIGS-DBs/FZCC0015.db
description ..................................: [Not found, but it's OK]
db_type ......................................: contigs (variant: unknown)
version ......................................: 24
DB Info (no touch also)
===============================================
project_name .................................: FZCC0015
contigs_db_hash ..............................: hashb19312d4
(...)
AVAILABLE GENE CALLERS
===============================================
* 'pyrodigal-gv' (1,436 gene calls)
* 'Ribosomal_RNA_23S' (1 gene calls)
* 'Ribosomal_RNA_16S' (1 gene calls)
AVAILABLE FUNCTIONAL ANNOTATION SOURCES
===============================================
* COG24_CATEGORY (1,223 annotations)
* COG24_FUNCTION (1,223 annotations)
* COG24_PATHWAY (497 annotations)
* KEGG_BRITE (1,028 annotations)
* KEGG_Class (324 annotations)
* KEGG_Module (324 annotations)
* KOfam (1,029 annotations)
If you wish to extend the phylogenomic analysis with your own contigs-db files, this is the time to add each genome you have into the CONTIGS-DBs directory, and extend the contents of this file with entries like this:
genome |
mapping_rep_accession |
subclade |
genus_species |
accession |
genome_extended |
genome_type |
is_95ANI_rep |
isolate_origin |
|---|---|---|---|---|---|---|---|---|
| MY_GENOME_NAME_01 | UNKNOWN | NEW_GENOME | ||||||
| MY_GENOME_NAME_02 | UNKNOWN | NEW_GENOME |
The genome name here should match to the name of the contigs-db you have put into the CONTIGS-DBs directory. If the name you put in into this file is MY_GENOME_NAME_01, your contigs-db file should be at CONTIGS-DBs/MY_GENOME_NAME_01.db.
Annotating contigs-dbs with Alphaproteobacterial SCGs
If you are here, it means you have all the contigs-db files in your work directory (with or without the contigs-db files you have for your own SAR11 genomes).
The next step is to prepare for phylogenomics, for which we needed to first determine which single-copy core gene (SCG) collection we should use. Previous studies, such as the one by Muñoz-Gómez et al (2019) that suggested that mitochondria are a sister clade to the Alphaproteobacteria, relied upon a curated set of 200 SCGs for Alphaproteobacteria. But not all those genes occurred in all SAR11 genomes, so we first evaluated the presence of the 200 SCGs alphaproteobacterial genes across our collection of genomes by using the anvi’o program anvi-script-gen-hmm-hits-matrix-across-genomes, and excluded genes that were missing in more than 90% of the SAR11 genomes we had in our collection. This resulted in a subset of 165 genes that were suitable for downstream phylogenomic analyses of this group. In our paper, we refer to this SCG collection as SAR11_165.
You can download the HMMs for SAR11_165 as an anvi’o hmm-source artifact from here to annotate our contigs-db files:
# download the HMM source to our work directory
curl https://merenlab.org/data/sar11-phylogenomics/files/Alphaproteobacterial_SCGs.tar.gz \
-o Alphaproteobacterial_SCGs.tar.gz
# uncpack it
tar -zxvf Alphaproteobacterial_SCGs.tar.gz
# get rid of the archive file
rm -rf Alphaproteobacterial_SCGs.tar.gz
We can now annotate all contigs-db files with these single-copy core genes using the following for loop:
for contigs_db in CONTIGS-DBs/*.db
do
anvi-run-hmms -c ${contigs_db} \
-H Alphaproteobacterial_SCGs \
--num-threads 8
done
Obviously, you can also do it on your HPC. But once you are done, the output for the following command in your work directory,
anvi-db-info CONTIGS-DBs/FZCC0015.db
should include a line like this:
DB Info (no touch)
===============================================
Database Path ................................: CONTIGS-DBs/FZCC0015.db
description ..................................: [Not found, but it's OK]
db_type ......................................: contigs (variant: unknown)
version ......................................: 24
DB Info (no touch also)
===============================================
project_name .................................: FZCC0015
contigs_db_hash ..............................: hashb19312d4
(...)
AVAILABLE HMM SOURCES
===============================================
* 'Alphaproteobacterial_SCGs' (165 models with 165 hits)
(...)
Phylogenomic analysis
For a phylogenomic analysis we will use the standard anvi’o phylogenomics workflow with an external-genomes file.
You can create an external-genomes-txt file for all your genomes using the program anvi-script-gen-genomes-file file:
anvi-script-gen-genomes-file --input-dir CONTIGS-DBs/ \
-o external-genomes.txt
If you included x number of new contigs-db files (or FASTA files) into your analysis, running the following command,
wc -l external-genomes.txt
Should give you 494 + 1 + x (that 1 comes from the header). This is your final check to make sure everything is in order, so if you are getting a number here that you don’t expect to see, please make sure you figure out what went wrong before moving onto the next steps of the analysis.
OK. If you are here, all is set.
Which means, we now can get the aligned and concatenated alphaproteobacteiral SCGs across our genomes using the program anvi-get-sequences-for-hmm-hits (please note that this step should take some time, so you can go get a coffee or a long walk that takes about 40 minutes (it is muscle aligning 480+ genes 165 times)):
anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt \
-o SAR11_165_concat.fa \
--hmm-source Alphaproteobacterial_SCGs \
--return-best-hit \
--get-aa-sequences \
--concatenate
Then we will use trimAL (already installed in your anvi’o environment) to trim our alignment so the positions that are gaps in more than 50% of the sequences are gone goodbye:
trimal -in SAR11_165_concat.fa \
-out SAR11_165.fa \
-gt 0.50
If you are here, it means you have your final FASTA file to run the phylogenomic analysis.
For this, we will use IQtree (also already installed in your anvi’o environment), a robust maximum likelihood algorithm to reconstruct trees. So regardless how you came all the way down here, you need to run this command on your HPC (with 40 cores in this particular example, that will still take over 40 hours to run):
iqtree -s SAR11_165.fa \
-m LG+F+R10 \
-T 40 \
-ntmax 25 \
--alrt 1000 \
-B 1000
After what feels like eternity, you will get a consensus tree from this analysis. Here is ours, which is neatly renamed to something that makes more sense:
https://merenlab.org/data/sar11-phylogenomics/files/sar11.tree
The difficult part is now done.
Visualization in anvi’o
There are indeed many ways to visualize phylogenetic trees. Everybody knows that. What might be something that is not well-known by many is that, anvi’o is quite a good to visualize them, as well. According to some, it is actually the best if you can believe that. Well, our study does not take sides in this debate. However, for the sake of convenience, here this tutorial will demonstrate how we visualized our tree along with the contextual information.
If you wish to start from scratch, you can simply download the final phylogenomic tree into your work directory,
curl https://merenlab.org/data/sar11-phylogenomics/files/sar11.tree \
-o sar11.tree
And immediately visualize it using the program anvi-interactive in what we call ‘manual’ mode:
anvi-interactive -t sar11.tree \
-p sar11.db \
--manual
This command will automatically generate a blank profile-db, and will open your browser to show you this once you click ‘Draw’:

Unedited, unrooted, raw tree
You can click any branch on your interactive display by pressing command or control key to re-root the tree,

Being a deity and choosing a branch to root the tree.
And even change the display mode to ‘phylogram’,

Switching from a circular view to a conventional phylogram view
And set some reasonable values for the display height and the width of the tree from the options panel,

Adjusting tree display width and height
To get to a display like this:

A more reasonable display
Finally, one can save the ‘state’ of the visual settings by clicking on the ‘Save’ button on the very bottom-left of the Settings panel, so everytime you run :

The very nicely hidden save state button
Of course we are not done here. The most important thing is to be able to visualize all the branch data and other key information for us to understand this organization, and/or display new genomes we have added to the mix together with all the others. Luckily, we have the genome-data.txt file in our work directory. Adding that information to this display is quite straightforward. Go back to your termianl, press CTRL+C to terminate the running instance of anvi-interactive, and re-run it with the genome data:
anvi-interactive -t sar11.tree \
-p sar11.db \
--additional-layers genome-data.txt \
--manual
This time, you will be looking at this display in your interactive interface:

Still ugly, but much more informative display
The reason this looks so busy is that because anvi’o displays every single column in the genome-data.txt file with whatever means possible. Some of them (such as ‘genome extended’) are not really useful to display. Others, such as the ‘genus species’ layer) should be text, etc. One can adjust all those through the ‘display’ subsection of the settings panel:

Unedited display properties for layers
Here is the lightly edited and reordered display properties,

Edited display properties for layers
And the result after re-setting clade colors and other minor adjustments:

A much more reasonable visualization of the tree and associated data
Much more reasonable, indeed. It shows less, but the information from the hidden layers are not lost, and you can see all the underlying data for each genome in the ‘data’ panel for your interactive exploration, or bin :

Mouse-over data display
If you like, you can click on branches to create bins, so there is even more information on the display before calling it final:

A much better and informative display
As you can imagine, if you have included FASTA or contigs-db files for your new genomes into this analysis at the beginning, this display with its search functions will help you to locate your genomes, and communicate their locations in any presentation or manuscript.
Once you are satisfied with your display, you can download the SVG file from the very bottom-left corner of the display, and finalize it for publication using any vector graphics tool. For our figures we used Inkscape.
Just for transparency and reproducibility, what you see in this screen is also avaialable as a data pack, and you can download and visualize it on your computer even if you haven’t done any of the steps in this tutorial the following way:
# download the data pack for interactive visualization of the SAR11
# phylogeny with all genomes
curl https://merenlab.org/data/sar11-phylogenomics/files/interactive-display.tar.gz \
-o interactive-display.tar.gz
# uncpack it
tar -zxvf interactive-display.tar.gz
# get rid of the archive file
rm -rf interactive-display.tar.gz
# go into the directory
cd interactive-display
# run the anvi'o interactive
anvi-interactive -t sar11.tree \
-p sar11.db \
--additional-layers genome-data.txt \
--manual
And voila (hopefully)!
Reproducing the same display from the data pack
Final words
We hope this was useful to you. Please feel free to let us know if you need any help.