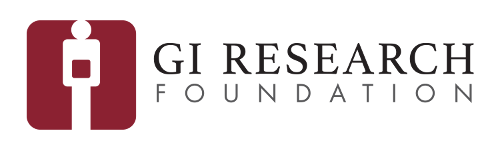Research
Table of Contents
Study hard what interests you the most, in the most undisciplined, irreverent, and original manner possible.

The vast majority of life on our planet is microbial. An astonishing number of microbial organisms living in terrestrial and marine habitats represent a biomass that exceeds every living organism that can be seen by naked eye, combined.
Through their inconceivable diversity that allow them to synthesize or break down a wide array of chemical substrates, microbes govern biogeochemical cycles that make Earth a habitable planet for much less talented organisms (such as ourselves). Our own body is also home to a diverse assemblage of microbial cells: bacteria that colonize our gastrointestinal tract help us maintain our health by extracting energy from undigested carbohydrates, synthesizing vitamins, and metabolizing xenobiotics.
Microbes are essential to Earth’s functioning at every scale. From comprehensive insights into ecosystem functioning to implementing effective bioremediation strategies or developing accurate models of environmental change and its long-term impacts, a detailed understanding of microbial life is one of the most critical requirements of environmental science and medical microbiology.
If you would like to learn more about the basic principles of microbial life, you should watch the first episode of the Microbial ‘Omics Seminar Series, a collaborative outreach effort our group organized in 2020 (and here is a bit of coverage of the event by Elise Wachspress).
Our research program focuses on understanding the ecology and evolution of naturally occurring microbial life using integrated ‘omics strategies and laboratory experiments: we develop new computational approaches and create advanced software platforms that intend to generate hypotheses from complex environmental data to bring us closer to explain mechanisms by which microbes interact with their surroundings, evolve, disperse, and initiate or adapt to environmental change.
We are environment agnostic as microbial ecology and evolution happens everywhere. Hence, our studies range from the human gastrointestinal tract and oral cavity, to sewage systems, insect ovaries, and oceans (OK, perhaps we find oceans just a little more intriguing than other habitats we study).
Describing our scientific interests as distinct categories is difficult since our studies are often intertwined with each other. Nevertheless, the following sections represent a crude and incomplete summary of our current interests.
Microbial ecology and evolution
Despite their great importance for the habitability of the planet and maintenance of our health, our understanding of the diversity, functioning, and evolution of microbial life is far from being complete (yes, despite all the probiotics you can buy from your local grocery store).
This is partially due to our inability to bring microbial life into the lab environment for comprehensive, conventional investigations: in most cases, it is very challenging to successfully isolate individual microbes from their interactive and complex environments and be able to learn about their natural behavior in controlled settings. And even when isolation is possible, understanding how well the isolated members of microbial populations represent their environmental population is not necessarily always clear. That’s why we complement conventional microbiological approaches with state-of-the-art molecular and computational strategies that allow us to study naturally occurring microbes to understand their lifestyles and evolution without having to bring them to the laboratory environment. That said, we do recognize the critical importance of being able to work with microbes in isolation and our fundamental need to generate mechanistic insights and see our science only complementary to such grand efforts.
Our group typically relies on cutting edge computational strategies and high-throughput sequencing data to identify known and novel populations of microbes, reconstruct microbial genomes from metagenomes, characterize their ecology, functional and metabolic potential, and/or characterize their population genetics.
For instance, one of our recent studies focused on SAR11, which is one of the most abundant microbial lineages in marine habitats. In collaboration with Stephen Giovannoni from the Oregon State University, Mike Rappé from the University of Hawai’i, and Ismail Uysal from the University of South Florida, we developed and applied novel approaches to investigate genetic diversity that emerges in a single SAR11 population as it travels around the world through the conveyor belt of large oceanic currents. By integrating metagenomics with in silico protein biochemistry, we were able to start generating hypotheses regarding the evolutionary processes that shape the proteome of this microbial clade as a function of changing ocean temperatures. Our ongoing efforts to explore strategies to further integrate microbial population genetics with protein structure will yield new powerful tools to generate testable hypothesis regarding the determinants of microbial fitness and evolution.
Deep-learning-estimated biogeography of the SAR11 subclade 1a.3.V based on single-amino acid variant profiles of its environmental populations, and example single-amino acid variants displayed on protein structures.
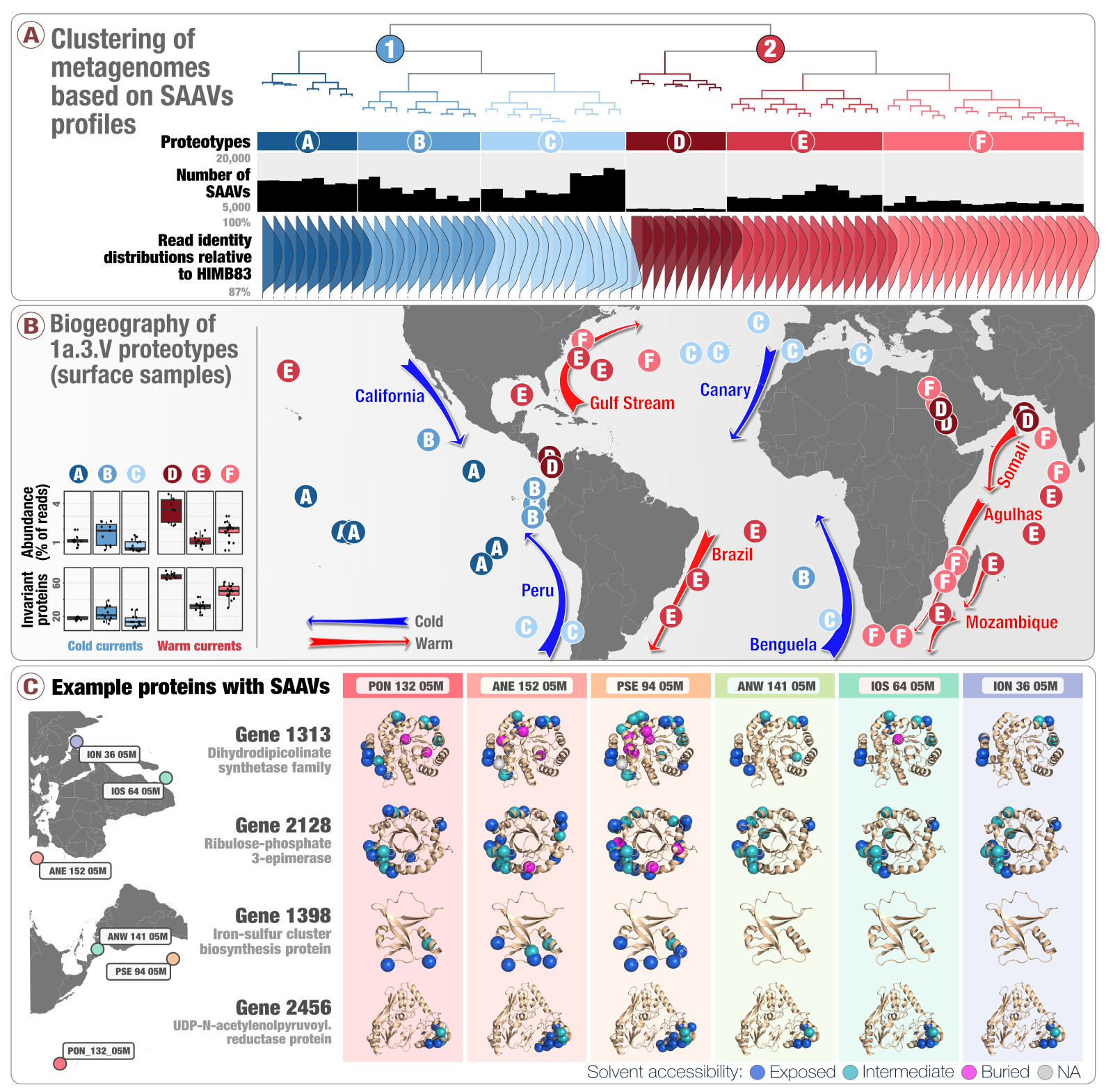
We also focus the powerful beam of integrative ‘omics to host-associated microbes, such as those that live in the human oral cavity. Oral cavity is a fascinating environment with multiple distinct niches in a relatively small space with no dispersal limitation: it has diverse anatomy with hard and soft tissue structures; it is under differential influences of the host immunity throughout the oral tissue types; and it is constantly exposed to exogenous factors. Oral microbes complement the richness of the oral cavity with their own sophisticated lifestyles: they form complex communities that show remarkable patterns of horizontal and vertical transmission across humans and animals, temporal dynamism, spatial organization, and site specificity. Altogether, the oral cavity offers a powerful environment to study the ecology and evolution of microbial systems, with which our group has a history. In collaboration with Jessica L. Mark Welch of the Marine Biological Laboratory, Amy D. Willis of University of Washington, Floyd E. Dewhirst of Forsyth Institute, and others, we recently integrated genome-resolved metagenomics, pangenomics, and phylogenomics to shed light on functional and genetic underpinnings of niche specificity of closely related microbes, and revealed that dental plaque may have served as a stepping stone at least for some environmental microbes to adapt to host environments.
Yes, it is annoying to have to brush off a piece of ‘outside’ in your mouth multiple times a day, but how cool it is to think that your dental plaque may have contributed to the evolution of environmental microbes to adapt host habitats? A phylogenomic analysis of new TM7 genomes we have reconstructed from human oral cavity shows that TM7 from dental plaque group together with environmental TM7, while tongue-associated TM7 group together with lineages associated with animal gut, suggesting that at least for TM7, the dental plaque resembles non-host environments, while the tongue and gut TM7s are more strongly shaped by the host.
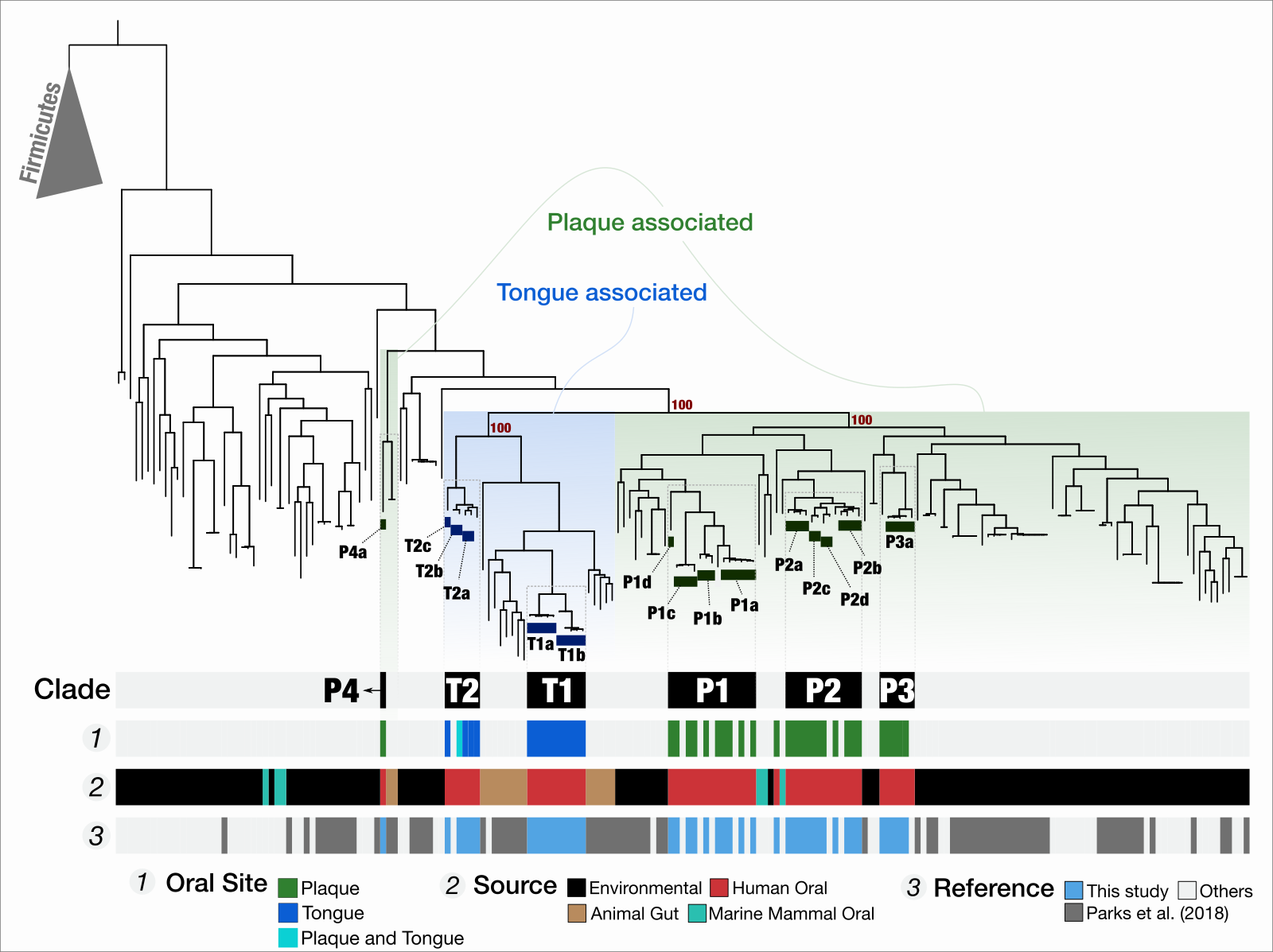
We are also interested in the human gut microbiome. During the last few years we have been leveraging Fecal Microbiota Transplantation (FMT) experiments, the quite literal transference of fecal matter from a healthy donor to a recipient, to study microbial ecology and evolution. FMT gained recognition as an effective treatment for recurrent or refractory Clostridium difficile infection (CDI). In addition to its medical relevance, we believe FMT is a great tool to gain deeper insights into microbial life as it opens a window into the event horizon of what happens when two microbial ecosystems collide.
The distribution of metagenome-assembled microbial genomes across FMT donor and recipients, as well as in healthy gut metagenomes across the United States shows that microbes populations that are good at colonizing unrelated individuals are also prevalent across health people!
Our group has been using FMT to study the functional basis of human gut colonization, but also to study microbial responses to environmental stress.
Microbial responses to environmental change
We imagine that in a hyper-dimensional space where every axis represents one of many distinct ecological factors (such as temperature, pH, light availability, the level of inflammation, etc), functions encoded in a single genome maps to a precise set of coordinates that define the niche boundaries of the organism the genome serves. But the Earth environments are not static. Thus, members of life are in a constant chase of an elusive equilibrium in their livelihoods they have evolved to occupy, as change constantly alters the fitness requirements of an environment by shifting the coordinates to which it maps. In other words, environmental change challenges the members of life to either go extinct, or find a way to change with it.
Some environments change more rapidly compared to others, others change relatively slowly. Some changes are complex and impact many axes of a given environment (such as temperature, which influences many of its co-variables), other changes are relatively simpler and impact a smaller number of dimensions (such as antibiotics). Change is not a single phenomenon either, but a nested set of phenomena spread through different scales of time and space that comes with a broad range of magnitude and dynamism.
Some ways for life to meet the challenge of change is through conventional means of evolution: a never-ending act of trial-and-error that changes the genetic code in small ways to move from one point in the fitness landscape to another. Our group studies this form of evolution through microbial population genetics and phylogenomics (where our work with marine microbes and residents of the oral cavity serve as examples). But environmental change is rarely so charitable to give enough time for life to adapt by perfecting its genetic code in a slow pace. Hence, our group also studies mechanisms by which microbes respond to environmental change in much smaller time scales by focusing on other means to adapt.
Some of the well-understood strategies to gain rapid fitness include horizontal gene transfers and acquiring plasmids, hypervariability through diversity generating retro-elements, and/or hypervariable genomic islands that emerge from constant shuffling of genes. But the fastest way to respond to respond to environmental change is translational regulation, which represents a profoundly under-explored layer of biology: protein translation dynamics that regulate fitness beyond the heritable genetic code.
A plethora of signal regarding how living cells respond to rapid changes in the environment lies at the level of translational regulation, a set of mechanisms by which members of all three domains of life can (and do) change their proteome in a plastic fashion and beyond heritable and expressed genetic information. If the genome is the cookbook and the ribosome is the chef, the epi processes that regulate the translation dictate how and when the chef deviates from the recipe. Transfer RNAs (tRNAs) are critical components of all living cells and properties of tRNA transcripts (i.e., their abundance, charging, and chemical modifications) serve as proxy to such epi processes. Our group has been collaborating with Tao Pan of University of Chicago to conduct systematical studies of tRNA transcripts beyond model organisms, and apply them to naturally occurring habitats by addressing significant molecular and computational bottlenecks. Through a three-year collaborative effort, we were able to demonstrate a novel workflow for direct sequencing and analysis of tRNA transcripts from naturally occurring microbes.
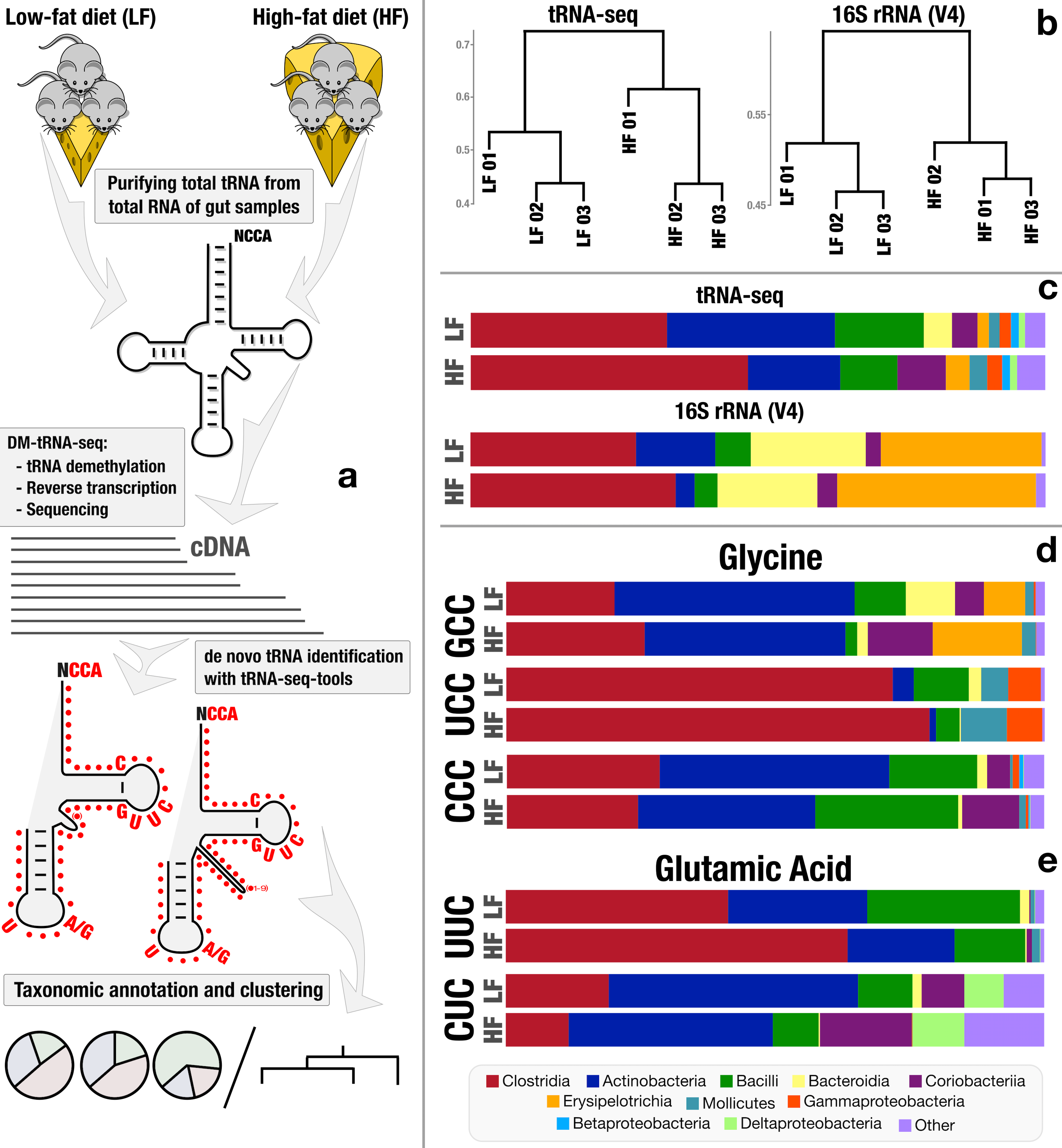
Draft tRNA-seq workflow prior to many biochemical advances made by the Pan lab over the last few months and computational advances made by Sam Miller, PhD, in our group.
Our proof-of-concept study showed that chemical modifications in tRNA transcripts were associated with protein translation dynamics linked to codon signatures that occurred more frequently in differentially expressed proteins in different environmental conditions. This unexpected discovery led to an award from the Keck Foundation award propelled us to further investigate translational dynamics in natural environments to accurately monitor subtle microbial responses at timescales and resolutions that reflect the speed of change in our oceans and terrestrial habitats. Our current goal to create a framework that gives access to tRNA sequencing in conjunction with other ‘omics strategies as we believe metaepitranscriptomics will likely influence our understanding of microbial ecology and evolution in fundamental ways, and help us to accurately model the interconnection between rapidly changing habitats and their inhabitants.
Advanced open-source software for high-resolution microbial ‘omics
Studying microbial life by integrating multiple ‘omics strategies required us to develop advanced software solutions such as anvi’o. We have implemented anvi’o as an open-source community resource.

Anvi'o is an open-source platform with more than 90,000 lines-of-code. Which really is a lot of code.
Computation is at the core of every scientific discipline. The fields of microbiology and microbial ecology, which rely on big data more and more, have also dramatically benefited from the advances in computation during the last decade. The importance of computation in life sciences puts our lab in a lucky situation, but we try to use our skills in computation wisely.
How much of the scientific questions we dare to ask depend on the availability of computational solutions that can facilitate the investigation of those questions in complex datasets we plan to generate? Although the inherent link between the tool and thinking will continue to bind the two together, we believe it must be mostly the intellectual curiosity what drives the direction of science, and not the comfort of what is available.
The agreement we have with ourselves in our group is to keep the biology as the sole inspiration of our direction, and never let the computational conveniences find their ways into our thinking in the expense of our ability to explore fundamental questions.
We intend to maintain our flexibility, and let the incoming questions shape and re-shape our software.
Our 2 cents
Some vocabulary we try to use, and promote as much as we can (for more, see our microbial ‘omics vocabulary):
-
“Bacteria and archaea” to describe the two
majorfamousdomainswhatevers of life, instead of “prokaryotes”. Because major scientific advances of recent times should not be ignored by scientists. Here is an opinion piece on this from Norman Pace: “It’s time to retire the prokaryote”. -
“Single-nucleotide variant” (SNV) to describe nucleotide positions with variation that emerges from the mapping of short reads from environmental shotgun metagenomes to a genomic context, instead of “single-nucleotide polymorphism” (SNP). SNP has a very specific definition, which makes it inappropriate to use in the context of metagenomic mapping.
-
“Metagenome-assembled genomes” (MAGs) to describe bins of assembled contigs from shotgun metagenomic data, instead of “draft genomes”. Because the term draft genome have long been used to describe not-yet-finalized genomes acquired from the whole genome sequencing and assembly of cultured microbial isolates.
-
“Marker gene amplicons” to describe the high-throughput sequencing data of marker genes targeted by (universal) primers, instead of “metagenomes”. Metagenomics is broadly defined as the study of genetic material directly recovered from a sample. With some effort, this broad definition could include marker gene amplicon surveys, but it really should not. The authors who coined the term associate the ‘metagenome’ with having access to the collective genomes of microbes in an environment. Today the term metagenomics is mostly used to describe shotgun sequencing of the environmental DNA in order to explore the functional potential or community composition of a given community at the level of metagenomic short reads (without assembly), assembled contiguous DNA segments (after assembly), or metagenome-assembled genomes (after assembly and binning). Marker gene surveys, including the ones that amplify hyper-variable regions of the ribosomal RNA genes, does not fit into what metagenomics describes, and the misuse of the term creates a lot of confusion.
-
“kbp”, “Mbp”, or “Gbp” to communicate the number of base pairs in a contig, or a genome, instead of “KB”, “MB”, or “GB”. Because the latter are commonly used to describe the amount of digital information in ‘bytes’, and the alternative use is not appropriate. Although it appears in the literature quite often, the use of “Kb”, “Mb”, or “Gb” are not very suitable either, since these units are commonly used to quantify digital information in ‘bits’.
-
Anything else to describe microbes that are not yet characterized, instead of “microbial dark matter”. Just stop it. Please.
If you believe we need corrections, please don’t hesitate to write to us :)
Thanks
Since the inception of our group at the University of Chicago, we were supported by the University start-up funds, private donors, foundations, and government agencies. We are very thankful for their trust.