
Table of Contents
Probably because the underlying logic behind oligotyping is remarkably different from the canonical approaches to demarcate high-throughput sequencing datasets, certain things about oligotyping sounds counter intuitive at first. This may be why I and others who are familiar with oligotyping hear similar questions from different audiences.
For instance, this statement seems to take some time to fully appreciate:
Oligotyping does not require alignment if the length variation across amplicon sequences is not artificial.
At this point I admit that this is not clear for everyone. I hope this post will help clarifying some of the question marks.
First and foremost, please read this answer first if you are asking yourself “when should I align my reads? when should I trim them, or when should I simply pad shorter ones with gap characters without alignment”:
https://groups.google.com/d/msg/oligotyping/KVcWdR_iWmA/qr5JXeKnNEgJ
For the lazy, here is a copy-paste from that post to address common cases. First, for the Illumina case (as you will see alignment is not necessary under no circumstances for Illumina):
-
Case 1: “I have Illumina data, all of my reads are equal in length”: You don’t need to do anything, you can just oligotype your dataset or analyze with MED.
-
Case 2: “I have Illumina data, length of my reads vary, because the quality filtering I used trimmed them at random locations”: Remove 10% of the shortest reads from your dataset, trim the remaining reads to the shortest one left in the dataset before the oligotyping / MED analysis. Optionally remove all the reads that were trimmed by the trimmer, and probably you will still have a lot of reads per sample.
-
Case 3: “I have Illumina data, length of my reads vary, because I merged partially overlapping reads”: Pad your reads with gap characters before the oligotyping / MED analysis (you can use
o-pad-with-gapsscript to do this).
Second, for people who work with 454 data:
-
Case 4: “I have 454 data, my reads are not aligned, but they are equal in length”: Align them using PyNAST, because someone trimmed your reads in a meaningless way. Follow the instructions in Case 5.
-
Case 5: “I have 454 data, the quality filtering I used trimmed them from random locations”: Align them using PyNAST against GreenGenes template, use
o-trim-uninformative-columns-from-alignmentscript to remove columns of gap characters without any nucleotide information, then RE-TRIM the resulting alignment usingo-smart-trimprogram with--from-end(if you have your forward primer intact) or--from-start(if your reverse primer intact) to a have a nicely trimmed dataset before the oligotyping / MED analysis. If you are not sure whether you are doing the right thing, send an e-mail to Meren. -
Case 6: “I have 454 data, all my reads reach to the distal primer, and I trimmed them all from the primer location”: Align your reads using PyNAST, use
o-trim-uninformative-columns-from-alignmentscript to remove columns of gap characters without any nucleotide information, and you are ready to go.
Don’t be alarmed if you find yourself thinking of the Illumina case and still wondering “but what happens if there is a natural insertion or deletion that shifts all bases? how does oligotyping help me solve it without an alignment?”.
Here is an attempt to clarify this:
Oligotyping requires reads in a dataset to start at an evolutionarily homologous region (i.e., following a primer site), and contain equal number of characters (or end at an evolutionarily homologous region –if this part doesn’t make sense, please do not continue and read the previous answer carefully). Since the artificial insertion or deletion of bases is not a common type of error for Illumina platforms, length variation among reads do represent biological differences (assuming you haven’t used a quality-filtering approach that trims reads from arbitrary nucleotide positions to preserve high-quality parts of them). In this case oligotyping can make sense of your reads without an alignment step. Yes, even if they contain natural insertions or deletions.
Assume a dataset of two very closely related organisms that differ from each other by a single insertion at nth nucleotide position at the sequenced region. If the user performs the entropy analysis on this dataset following an alignment, there would be a single discernible entropy peak at position n, since all reads lacking the inserted base would be accommodated with a gap character at nth position. An oligotyping analysis using this position would result in two oligotypes, one of which represented by a gap character, and the other represented by the identity of the inserted base. On the other hand, if the user performs the entropy analysis on this dataset without an alignment, in this case there would be multiple peaks starting at nth position, spanning the remaining bases following position n, and each entropy peak would be equal in length when they are present. An oligotyping analysis using any of those entropy peaks (or any combination of them) would result in the same two oligotypes. Following figure is the translation of this paragraph into a visual form:
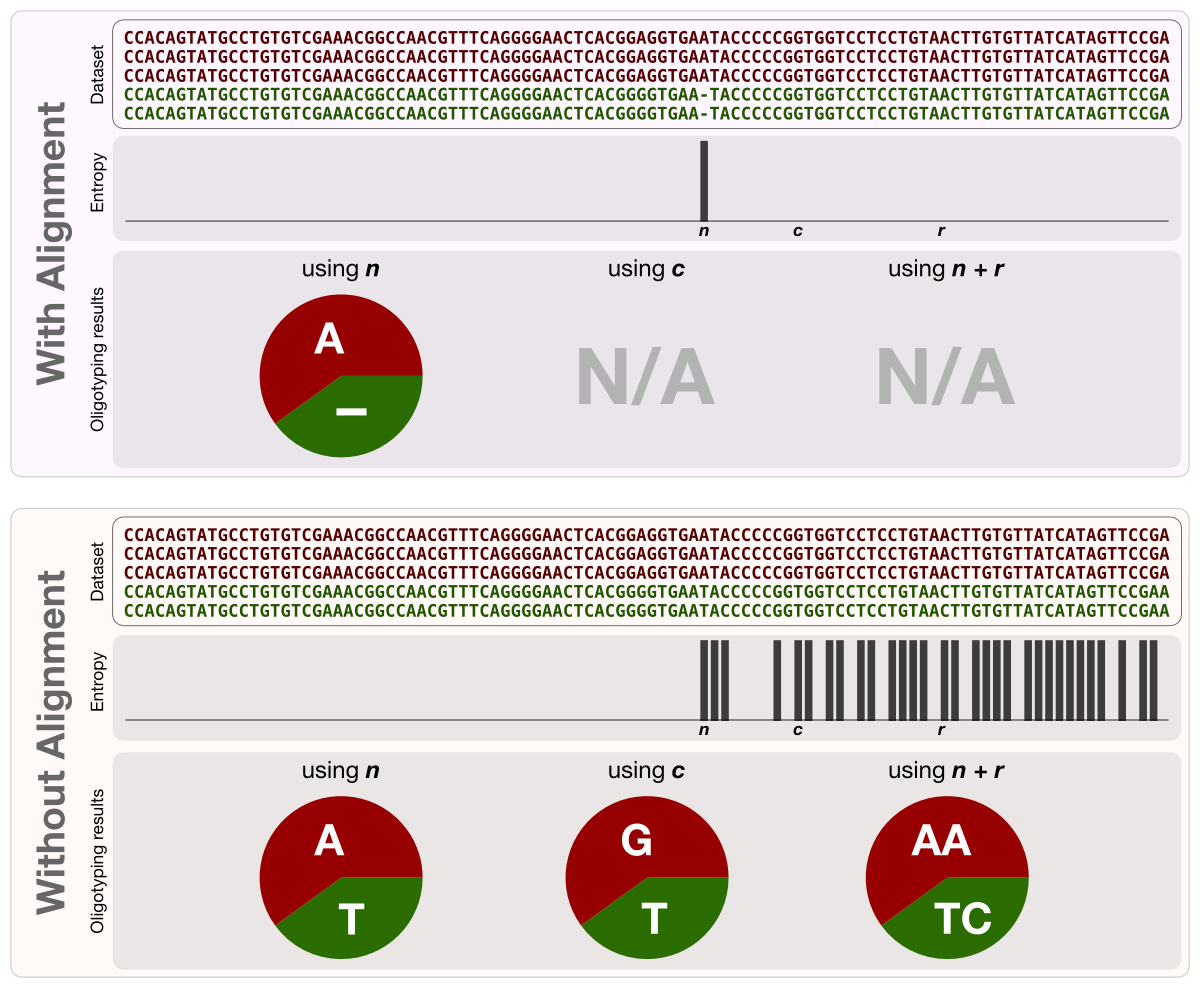
Each panel in the figure demonstrates an entropy analysis and the following oligotyping analysis of the same mock dataset with or without alignment. Below the entropy analysis results arbitrary nucleotide positions n, c and r are marked. Separate oligotyping analyses using these positions and recovered oligotype identities are shown as piecharts. As you can see, regardless of the chosen entropy peak(s) for oligotyping analysis, results (meaning the representative sequences and their relative abundances) are identical for the same dataset with or without alignment.