
Table of Contents
- Debating the shared history of Archaea and Eukaryota
- The archaeal origin for the eukaryotic life hypothesis: the genomic evidence
- Lokiarchaeum sp. GC14_75, Loki2 and Loki3
- A close look at Lokiarchaeum sp. GC14_75 in the context of coverage and single nucleotide variants
- Curating Lokiarchaeum sp. GC14_75 using sequence composition and differential coverageI
- Final notes
- Data availability
It has been in my to-do list for many months now, but I just finally was able to take the time to explore the composite genome Lokiarchaeum sp. GC14_75.
Meren’s pirate note: There are two videos in this post in which Tom kindly demonstrates the interactive capabilities of anvi’o. We know how much everyone likes videos, so here they are in case you don’t have time for the text: anvi’o inspection, anvi’o curation. Also the data to reproduce everything here is available at the very end of the post.
Lokiarchaeum sp. GC14_75 was recently proposed to bridge the gap between prokaryotes and eukaryotes, possibly shedding light on the origin of eukaryotic cells.
My inspection and curation efforts using the anvi’o interactive interface may be relevant to the ongoing debates. This blog also aims to describe, using a real-world example, the simple steps of inspecting metagenome-assembled genomes (MAGs) using anvi’o. These steps are critical not only to assess the quality of MAGs, but also to observe the complexity of metagenomic data, and by extension, the microbial world.
Debating the shared history of Archaea and Eukaryota
As everyone in life sciences know, the widely accepted Tree of Life organizes domains of life into three groups: Bacteria, Archaea, and Eukaryota (Woese and Fox, 1977). This observation was made possible by using molecules from distant life forms as documents of evolutionary history, a powerful concept introduced earlier by Zuckerkandl and Pauling (1965). The rapid growth in genomic discoveries from various environments, with or without the support of cultivation, reveals new bacterial, archaeal, and eukaryotic lineages at an unprecedented pace (e.g., Hug et al., 2016, Jungbluth et al., 2016, Hirashima et al., 2016) (Note: the previous sentence, which incorrectly cited Hug et al. publication as a support for the three domain model, has been corrected after Jonathan Eisen’s comment down below).
Nevertheless, the origin and evolution of these domains (and particularly the shared history between Archaea and Eukaryota) are not fully understood and represent an area of intense scientific debate (Gribaldo et al., 2010). This led to a situation where astrophysicists can describe in great details the chronology of the known universe starting 10-43 seconds after the big bang, but biologists cannot agree on the origin and main evolutionary paths of the life forms we interact with, here on Earth (which keeps getting warmer and warmer).
The archaeal origin for the eukaryotic life hypothesis: the genomic evidence
In 2015, Anja Spang, Jimmy H. Saw, Steffen L., Jørgensen, Katarzyna Zaremba-Niedzwiedzka, and others contributed to the debate by publishing genomic discoveries favoring the hypothesis of an archaeal origin for eukaryotes (Spang et al., 2015).
Briefly, the authors used assembly-based metagenomics to characterize the genomic content of members of a new archaeal phylum (Lokiarchaea) that is found in deep-sea sediments. Their metagenomic and subsequent phylogenomic analyses (i.e, phylogeny using a concatenation of multiple gene markers) indicated that this lineage represent the nearest relative of eukaryotes, thus bridging a gap at the genome-level between the two domains of life. More than one hundred research articles have already cited this study, and some of our peers see it as critical evidence supportive of a two-domains model. Due to the importance of the results, part of the scientific community has been re-analyzing the data from various angles.
A few concerns and re-interpretations of the original analysis have been summarized in a blog. One poster abstract presented by Violette Da Cunha in the 2016 SMBE conference provides some additional information. Finally Nasir et al. (2016) published an article just recently challenging claims made in the original study.
Here is short summary of concerns (this might not be exhaustive):
- Accuracy of the metagenomic-assembly outputs (chimeric genome / false discoveries)
- Relevance of concatenates genes (phylogenomics), compared to more standard phylogenetic tools (using a single phylogenetically-relevant gene).
- Bias in the set of taxa from the tree domains of life used in the study to link Archaea and Eukaryota.
Thus, it appears that the original interpretation of the results did not yet reach a consensus among the community. This triggered my curiosity, and I decided to explore some aspects of the data, with a focus on metagenomic binning and the interpretation of read recruitment. Especially, I tried to understand what really the (Lokiarchaeum sp. GC14_75, the only Lokiarchaea genome available so far, represents in the context of the metagenomic data produced in the original study.
Lokiarchaeum sp. GC14_75, Loki2 and Loki3
Binning genomes from metagenomes is the process of organizing metagenomic assembly results into biologically coherent units, i.e., metagenome-assembled genomes (MAGs). From their metagenomic data, Spang et al. recovered a relatively large Lokiarchaea MAG (Lokiarchaeum sp. GC14_75, >5Mbp) using a manual binning workflow.
They described their MAG as “a 92% complete, 1.4 fold-redundant composite genome”. Authors subsequently used the GC-content to resolve this MAG into two distinct groups (Loki2 and Loki3). Here is a screenshot of the figure corresponding to this analysis so everyone can have a better understanding of the information the GC-content holds in this particular example:
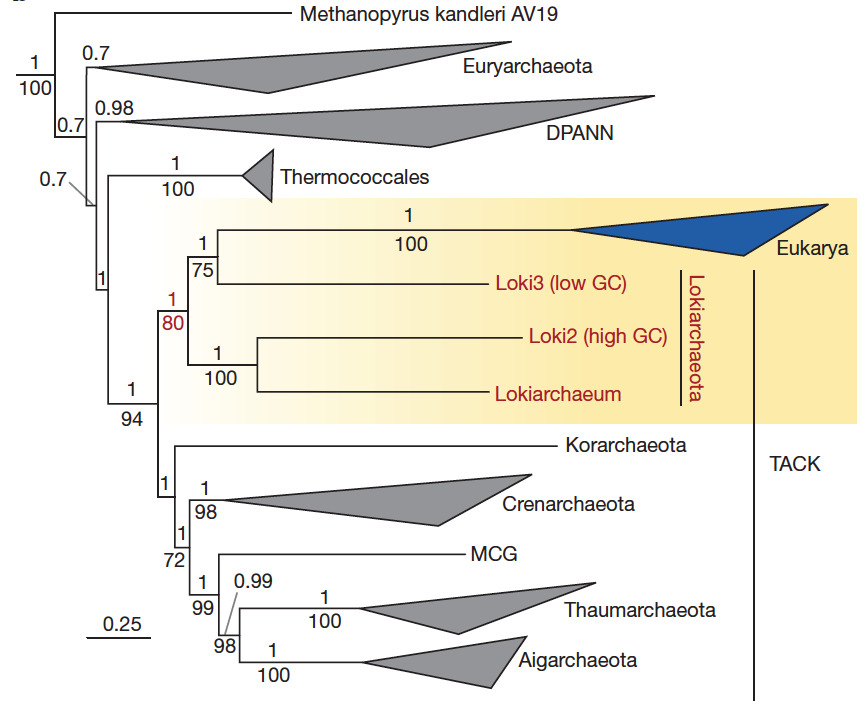
Loki2 and Loki3 have a GC content of 32.8% and 29.9%, respectively. Their completion and redundancy values were not described in the text, and the corresponding FASTA files have not been made publicly available. Yet, Loki2 and Loki3 represent a key component of the study and have been used to generated the phylogenomic tree(s). Here is a screen shot from the figure 2, panel b:
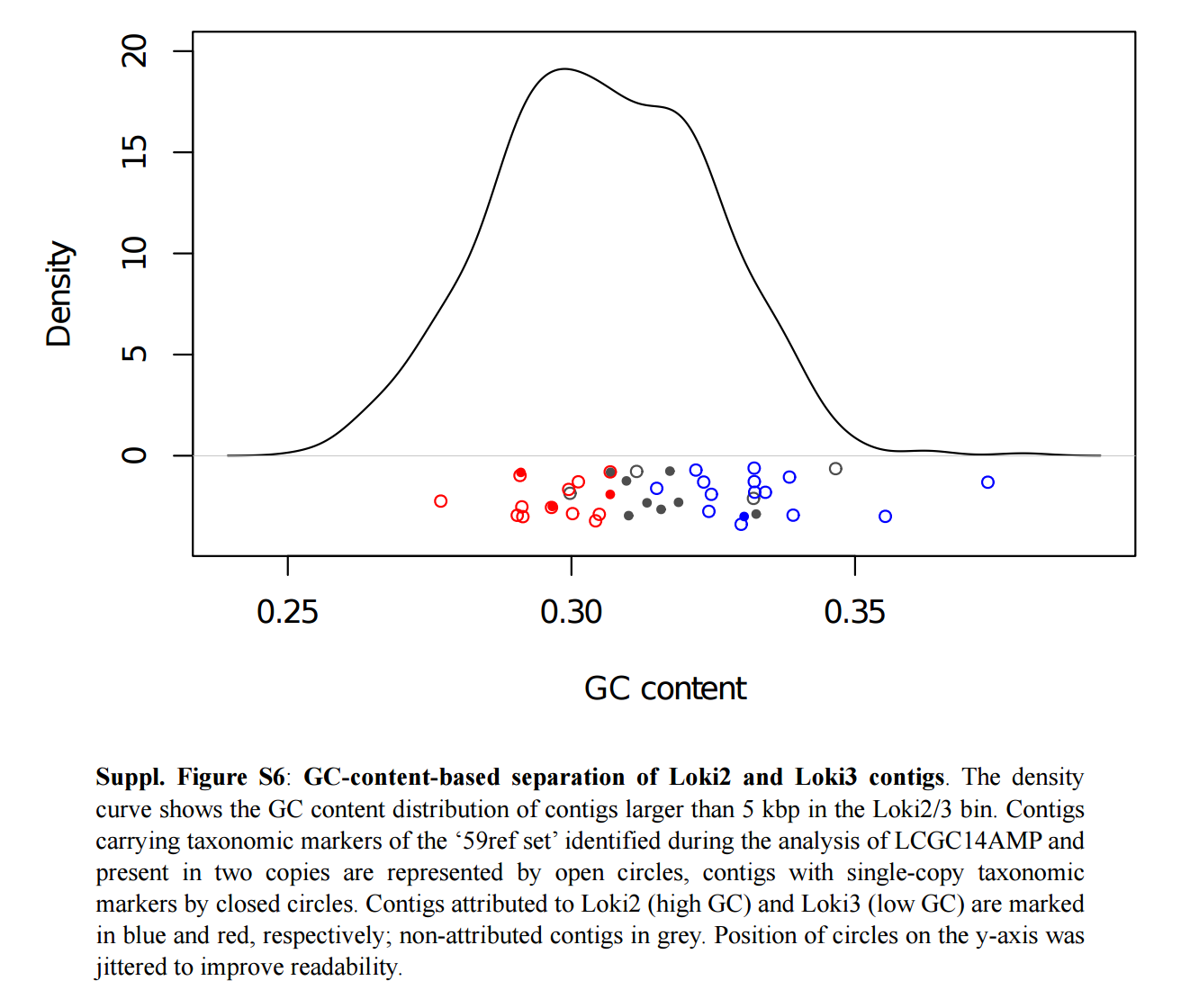
So, at this point the only Lokiarchaea genome available (the MAG Lokiarchaeum sp. GC14_75) is highly redundant and has only been resolved using GC-content. It is important to note that this variable is not commonly used for binning. Thus, the reasoning for a GC-content-based binning strategy is unclear, and might have had important consequences regarding the biological relevance of Loki2 and Loki3. So I decided to inspect and curate the MAG Lokiarchaeum sp. GC14_75 using anvi’o.
I first downloaded the MAG and the metagenomic reads used in the study (NCBI bioproject PRJNA259156; three SRA files), and then followed the anvi’o metagenomic workflow to process all files. Briefly, I generated a CONTIGS.db file from the MAG (after renaming contig headers appropriately), and searched for bacterial and archaeal single-copy gene collections using anvi-run-hmms. Anvi’o automatically identified the MAG as archaeal upon the occurrence of single copy genes, and determined a redundancy value of 56.8%, slightly above the value provided in the original study. I then used Bowtie2 to map metagenomic reads from the SRA files into the MAG and generate BAM files. I profiled the three BAM files separately using anvi-profile, and merged them using anvi-merge. The resulting anvi’o profiles to reproduce all analyses here are available at the end of the post.
A close look at Lokiarchaeum sp. GC14_75 in the context of coverage and single nucleotide variants
The anvi’o interface makes it easy to inspect contigs by visualizing coverage variations at the nucleotide level and the occurrence of single nucleotide variants (SNVs). This level of resolution is critical to assess the biological relevance of genomes, contigs and genes in the context of metagenomic data. I used anvi-interactive to manually inspect the contigs from Lokiarchaeum sp. GC14_75, and created a first short video to describe our exploration of the MAG:
I observed two important trends from this inspection effort:
- The occurrence of a high amount of SNVs across contigs
- The non-specific recruitment of metagenomic reads originating from multiple populations in regions often matching to one or multiple adjacent genes.
So, Lokiarchaeum sp. GC14_75 appears to be highly heterogeneous in the investigated samples, and contains genomic regions recruiting reads originating from multiple populations. In our humble opinion, both the unstable coverage regions, and the occurrence of SNVs should be considered in downstream phylogenomic analyses. But what about the redundancy issue and the biological relevance of Loki2 and Loki3?
Curating Lokiarchaeum sp. GC14_75 using sequence composition and differential coverageI
Both the sequence composition (e.i., tetra-nucleotide frequency) and differential coverage of contigs across metagenomes can be instrumental in resolving high-quality MAGs in metagenomes assembly outputs into high quality MAGs. Similar to my inspection effort, I created a second short video describing my binning strategy in the case of Lokiarchaeum sp. GC14_75 and the reasoning behind it:
Breifly, the organization of the investigated contigs upon differential coverage and sequence composition appeared to represent an effective clustering approach for binning. It provided two very distinct clusters with redundancy values below 7% that I will refer to as Lokiarchaeum 01 (GC-content of 31.21%) and Lokiarchaeum 02 (GC-content of 31.22%).
I also summarized the main results in here (for those too impatient to watch videos):
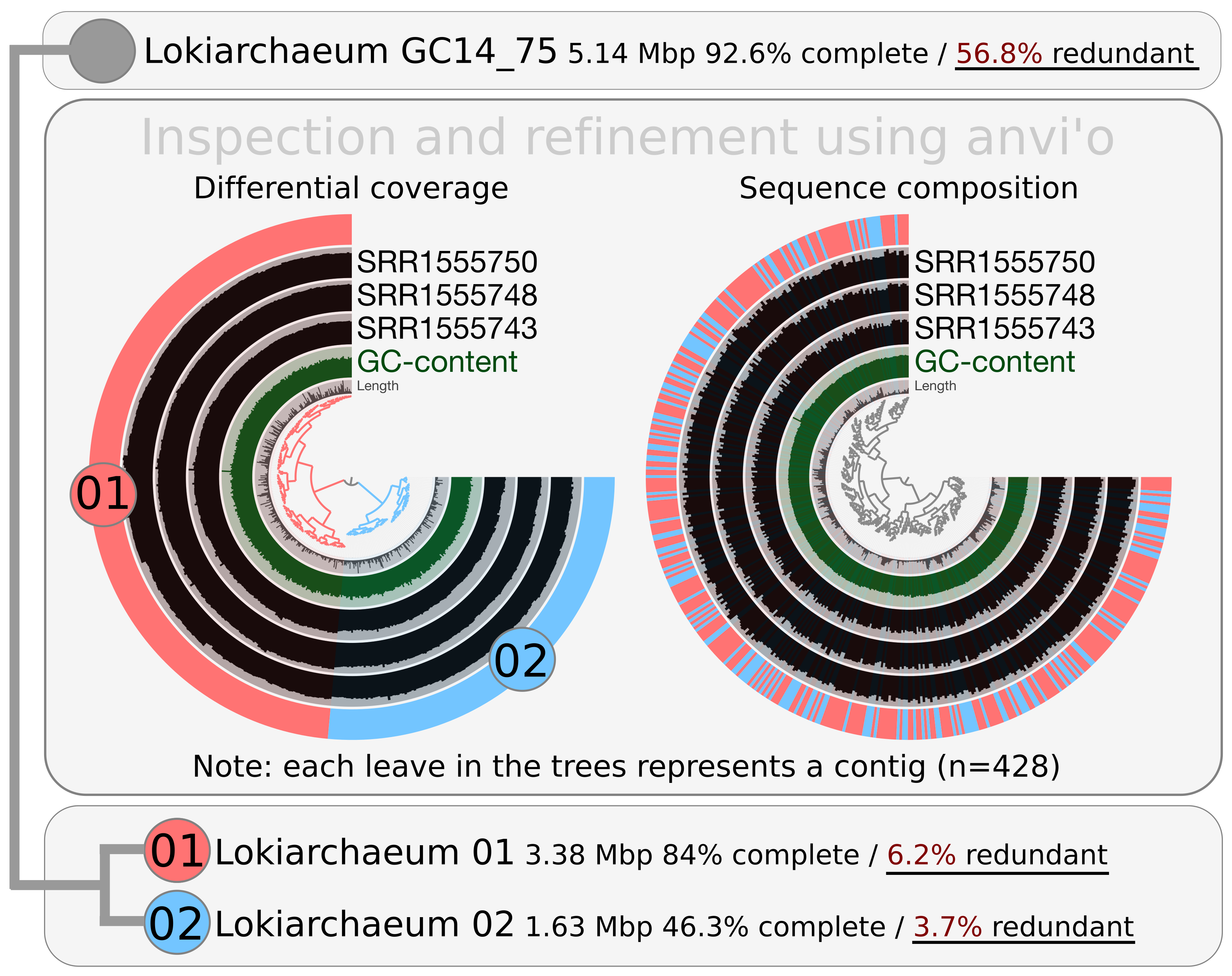
Lokiarchaeum 01 is larger than Lokiarchaeum 02, exhibits a higher completion value (84%), and was systematically more abundant in the investigated metagenomic dataset. The GC-content and sequence composition of the two MAGs was highly similar, suggesting they corresponded to closely related taxonomical lineages. Clearly, Lokiarchaeum 01 and Lokiarchaeum 02 represent very different binning outputs compared to what was performed in the original study. I do not claim that our binning strategy is better. However, differential coverage is considered more relevant than GC-content for binning (especially when sequence compositions are highly similar) and using this information resulted in two MAGs exhibiting relatively low redundancy values.
Final notes
What did we learn so far regarding the composite MAG Lokiarchaeum sp. GC14_75?
-
It contains a lot of SNVs when recruiting reads from the metagenomes it was characterized from. As a result, the consensus genomic sequence represents only a subset of the diversity present in the studied environment.
-
It recruited reads originating from multiple populations in specific genomic regions. Thus, these regions are likely not characteristic to a single population.
-
It is highly redundant and represents an amalgam of at least two populations with distinct relative distributions in the studied environment. I could resolve most of the redundancy by clustering contigs upon sequence composition and differential coverage. This led to the characterization of two MAGs named Lokiarchaeum 01 and Lokiarchaeum 02.
It is likely that Lokiarchaeum 01 and Lokiarchaeum 02 may still represent a bridge between Archaea and Eukaryota. But it would need someone to take a look at the phylogeny of these Lokiarchaeum MAGs, if possible by taking into account the SNVs and regions exhibiting a high amount of non-specific read recruitments.
Finally, I hope that after watching the two videos, some of you will consider using anvi’o to inspect and curate genomes in the context of metagenomic data. These steps are relatively easy, and might help avoiding yet another Tardigrade drama.
Data availability
All the files used here to analyze, inspect and curate the composite MAG Lokiarchaeum GC14_75 have been generated using anvi’o v2.1.0 and are publicly available:
-
Anvi’o files: Download the archive, unpack it, and run the interface by typing
anvi-interactive -p PROFILE.db -c CONTIGS.db. -
Static HTML summary: Download the archive, unpack it, double-click
index.htmlfile to access the FASTA files for the two curated Lokiarchaeum MAGs,Lokiarchaeum_01andLokiarchaeum_02.
Although writing the blog post took forever, the entire analysis (starting from the download of the genome and SRA files from NCBI to anvi’o profiling and selections) took about 4 hours.
Curating Loki archaea: https://t.co/J9yAEOrqIf. An insightful inspection effort with videos by @tomodelmont.
— A. Murat Eren (@merenbey) January 3, 2017