
Table of Contents
There is a cool pre-print out there:

The people who are responsible for this work include Mark Achtman, M. Thomas P. Gilbert, and Zhemin Zhou.
Briefly, the authors used shotgun metagenomics to recover a Salmonella enterica genome from a 800-year-old skeleton (already quite cool). They call this genome Ragna genome in the context of their study. After analyzing it with respect to 50,000+ modern S. enterica genomes in Enterobase as well as 100+ genomes they sequenced and assembled from the Institut Pasteur’s pathogen collection for better phylogeny, the authors suggest that the lineage to which Ragna genome belongs has been around as a human pathogen for at least one millennium, and it originated from a lineage of swine pathogens.
The study has everything. An exciting set of novel (from the corpse and from the Institut Pasteur collection) and publicly available (from Enterobase) data, an array of impressive methods to connect everything, and very interesting findings. Plus, it comes out as a pre-print first, and releases all of its data openly.
The methods section is here as a separate file, and everything in the pre-print is really neatly available. Including the supplementary data, representative genomes used for the pangenomic analysis, and most importantly the metagenomic short reads, from which they recovered 11 bacterial genomes, as well as the Ragna genome through read recruitment (for which they used modern relatives of Paratyphi C to identify sequences specific to the Ragna genome). Research groups that release the entire data behind their claims like champions, even when they can clearly get away without doing it, indeed deserve a bigger thanks from the community. So on behalf of the science community, I hereby thank Zhemin and his colleagues bigly.
One of the few impressive approaches in the study
If you are interested in metagenomics, pangenomics, or phylogenomics, you should consider going through this study. I also think it would make such a great journal club material. There are a number of very cool approaches in it, but one of the coolest things in this study from my subjective perspective is the fact that the authors have used DNA damage estimates to tease apart contaminant metagenomic bins from the ones they are interested in.
Probably it is quite well-known among the people who study ancient DNA, but I was utterly impressed by this technique thanks to my ignorance. So here is a bit more context for the ones who know about ancient DNA as much as I did until 15 minutes ago: The authors recover 11 near-complete bacterial genomes through metagenomic sequencing of the material from the corpse, assembling the resulting short reads into contigs, and finally organizing contigs into genome bins with CONCOCT. When you take a sample from a body that’s been buried for the last 800 years it is conceivable to have some environmental contamination in it. But how to determine which genomes are contaminants from the surrounding soil, and which ones are endogenous to the body? How can one learn about that? If you think about it this is quite a challenging question. But it seems there is a very smart way of solving this puzzle: deamination (which is a hydrolysis reaction that removes an amine group from cytosine and turns it into uracil). It turns out the passing of time takes its toll on DNA molecules through deamination. Which, in the context of this study, makes endogenous DNA contain more deaminated cytosines towards the end of each fragment, compared to the DNA fragments that comes from relatively recent day organisms. Isn’t this absolutely brilliant? No? You are not surprised? Well, the beauty of being a computational person in life sciences is that you get to enjoy things life scientists have learned probably when they were undergrads. While I was trying to better understand this phenomenon, I run into this lovely paper, in which the relationship between ancient DNA and deamination was probably shown for the first time. Here is a very good sentence from its abstract:
(...) Here we analyze DNA sequences determined from a Neandertal, a mammoth, and a cave bear. We show that purines are overrepresented at positions adjacent to the breaks in the ancient DNA, suggesting that depurination has contributed to its degradation. We furthermore show that substitutions resulting from miscoding cytosine residues are vastly overrepresented in the DNA sequences and drastically clustered in the ends of the molecules, whereas other substitutions are rare (...)
Yay for deamination of cytosine!
A side story on deamination Of course, just like everything in science, the reliance on deamination patterns is not straightforward. While I was learning about the topic, I came across a little debate between two groups around deamination. Pretty much this is how it goes:
- Group 1: This wheat is 8,000 years old.
- Group 2: No, it isn’t.
- Group 1: Yes, it is.
And here is a blog post from one of the members of the Group 1, where he, in my opinion, rightfully complains about the journal eLife. Just in case you needed some side stories to intensify your procrastination.
So, Zhemin and his colleagues determined (via mapDamage by Jónsson et al.) the deamination rate in genomic fragments of their 11 CONCOCT bins and the Ragna genome to set aside 9 of the 11 genomes as contamination. One thing that I didn’t get was the big difference between the estimated DNA damage for human contigs (0.47) and the estimated DNA damage for Ragna genome (0.9). Could it be due to the fact that chromosomes in eukaryotic cells are more protected from environmental stress compared to non-eukaryotic cells? Am I not making any sense? FINE…
A quick look at genome bins
As a small ‘thank you’ for releasing the data in an easily accessible form, I decided to volunteer myself for an independent review of the genome bins the authors released.
First I got the data (took about 1 minute):
curl -L -O http://wrap.warwick.ac.uk/85593/8/DatabaseS8-MAGs_from_CONCOCT.zip
unzip DatabaseS8-MAGs_from_CONCOCT.zip
ls *fa > bins.txt
sed -i '' 's/.fa$//g' bins.txt
Then I generated anvi’o contig databases for each bin, and run HMMs on them (took about 5 minutes):
for bin in `cat bins.txt`
> do
> anvi-gen-contigs.database -f $bin.fa -o $bin.db
> anvi-run-hmms -c $bin.db
> done
Then I took a look at the completion / redundancy estimates (took about 10 seconds):
for bin in `cat bins.txt`
> do
> anvi-script-get-collection-info -c $bin.db
> done
This is the result:
| CONCOCT bin | Percent completion | Percent redundancy | Num contigs | Total bases |
|---|---|---|---|---|
| Cluster1 | 89.92% | 3.59% | 919 | 3,509,680 |
| Cluster2 | 84.89% | 2.15% | 267 | 2,110,679 |
| Cluster3 | 76.25% | 1.43% | 1,042 | 1,991,946 |
| Cluster18 | 95.68% | 2.87% | 397 | 4,068,232 |
| Cluster25 | 86.33% | 1.43% | 897 | 1,897,048 |
| Cluster30 | 91.36% | 4.31% | 641 | 1,844,992 |
| Cluster32 | 93.52% | 8.63% | 1,113 | 3,052,702 |
| Cluster40 | 91.36% | 5.75% | 995 | 4,907,161 |
| Cluster66 | 77.69% | 2.15% | 1,981 | 3,794,852 |
| Cluster69 | 83.95% | 4.93% | 802 | 2,035,509 |
| Cluster72 | 90.64% | 2.87% | 470 | 1,319,922 |
Well, they look awesome to me. Then I wanted to quickly see how they looked like without any mapping. For this, I needed to generate some blank profile databases. So that’s what I did first (took about 30 seconds):
for bin in `cat bins.txt`
> do
> anvi-profile -c $bin.db -S $bin -o $bin --blank
> done
Then I was ready to visualize stuff in the anvi’o interactive interface (took about 5 minutes to inspect all of them and taking screenshots):
for bin in `cat bins.txt`
> do
> anvi-interactive -c $bin.db -p $bin/PROFILE.db
> done
Which displayed each contig in each CONCOCT bin one by one (the min/max boundaries for the GC-content are 0% and 75%):
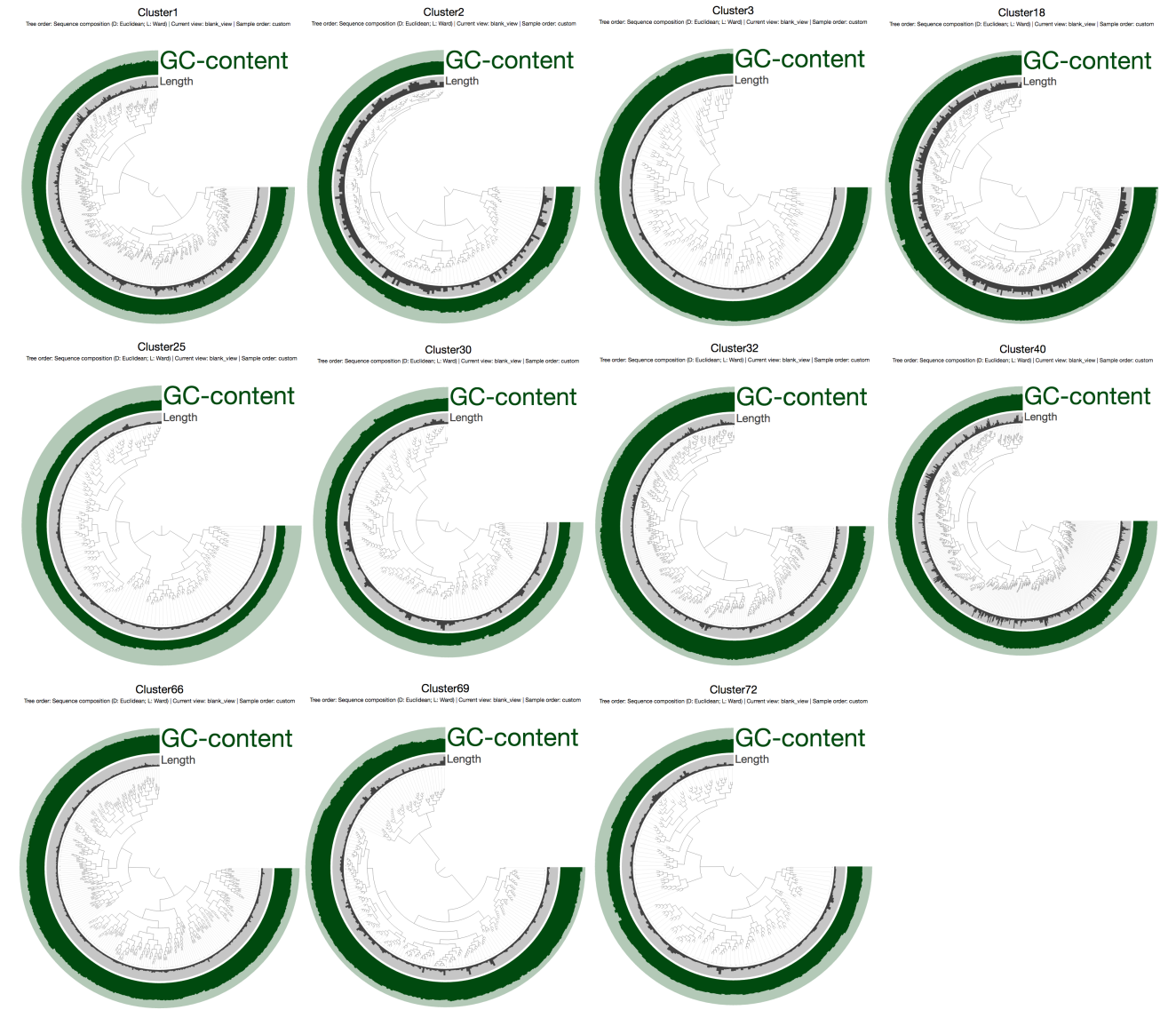
Of course without the mapping data it is a very coarse look. But things seem to be in order. I am always pleasantly surprised with the accuracy of CONCOCT (it does make mistakes, of course, but much less so than others). The only bin that somewhat looked as if it may have some contamination was Cluster 40, but a quick check in the interactive interface convinced me that it was probably OK, too:
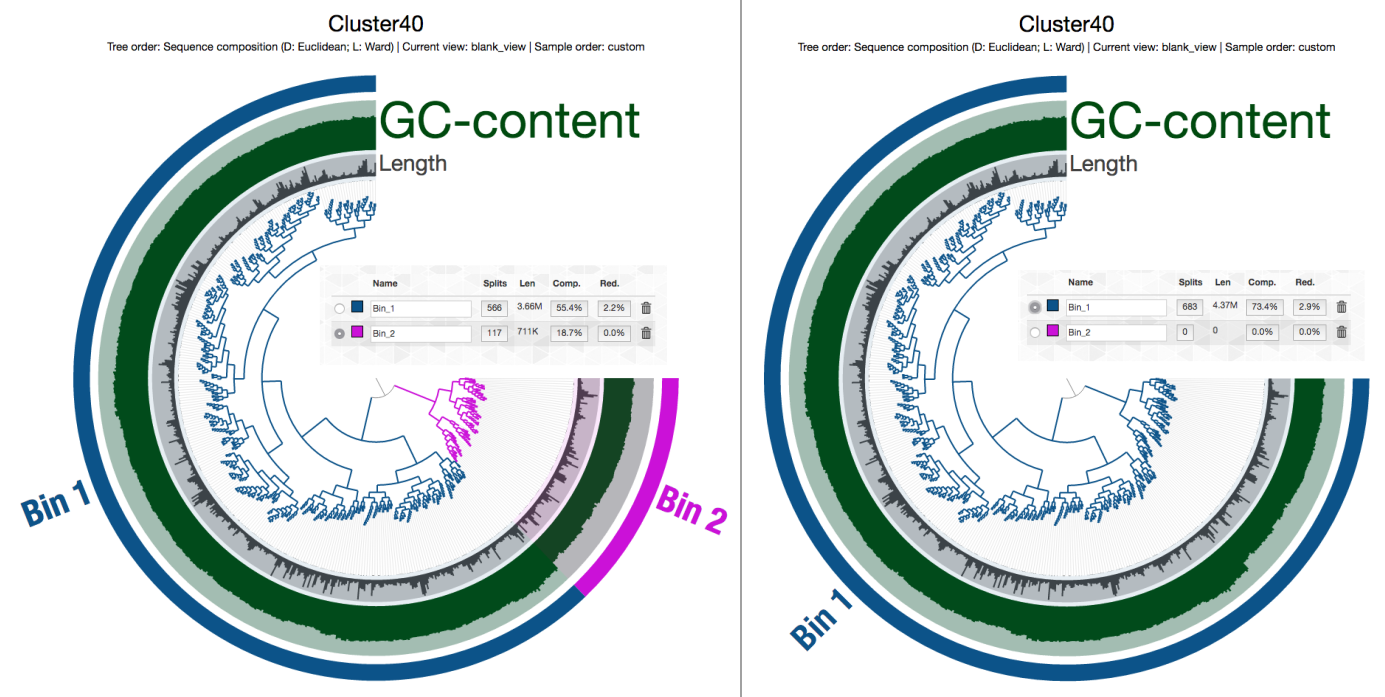
And then I did this, so you can download this entire directory from here, and play with it if you have anvi’o v2.1.0 or later:
cd ..
mv DatabaseS8-MAGs_from_CONCOCT Zhou_MAGs
tar -zcf Zhou_MAGs.tar.gz Zhou_MAGs/
The Ragna genome is not among these bins, but it is available via Enterobase. You can go to the Salmonella section in the Entorobase web site, and search for Ragna through the Search Strains link.
Of course without mapping, these analyses are not complete. But for these metagenome-assembled genomes, based on what I’m seeing so far, I am putting my 2 cents on the ‘approve’ button.
As of this beautiful Sunday evening, I am in the process of downloading the metagenomic data from here (I again thank the authors for their open science practice), and I will likely find ways to procrastinate more in the coming days to update the post with mapping results.
A lovely anvi’o figure
The rest of the pre-print is filled with very impressive stuff. Especially if you have been working with 16S data, and now slowly transitioning into metagenomics and/or comparative genomics, please do yourself a favor and carefully read this great work. Computational folk, relatively younger generation, and people who learned about the microbial world through marker genes have missed all these, and it is truly mind opening to see how much has already been done in microbiology before we came into it. I would have been happy to assume I am the only ignorant around here, but unfortunately I know that I am not.
I am definitely looking forward to reading this work in its published form.
A self-correction ~2 hours after posting this online: I wrote the sentence above, posted the blog online. Then Aaron Darling appeared in my mind out of nowhere, and said “it is already published, Meren, pre-prints are publications”. Yes, it is so true. Thanks, Aaron. But isn’t it funny how much we shoot ourselves in the foot by assuming that the status ‘published’ is some holy and magical designation that can only be assigned by journals we created by submitting our best work? Yes. No. This work is already published. Well, but it is not peer-reviewed, Meren. Fine, it is not yet peer-reviewed, but you know what, when I really think about it, I feel much better about it than I feel about some of the peer-reviewed work out there without any raw data. So that’s that. Here I apologize for unintentionally making it sound as if pre-prints are any lesser publications, and punish myself with ten push-ups. Proceeds to pay his debt.
Well, the real reason I decided to take a better look at this study by writing a blog post was in fact this gorgeous figure:
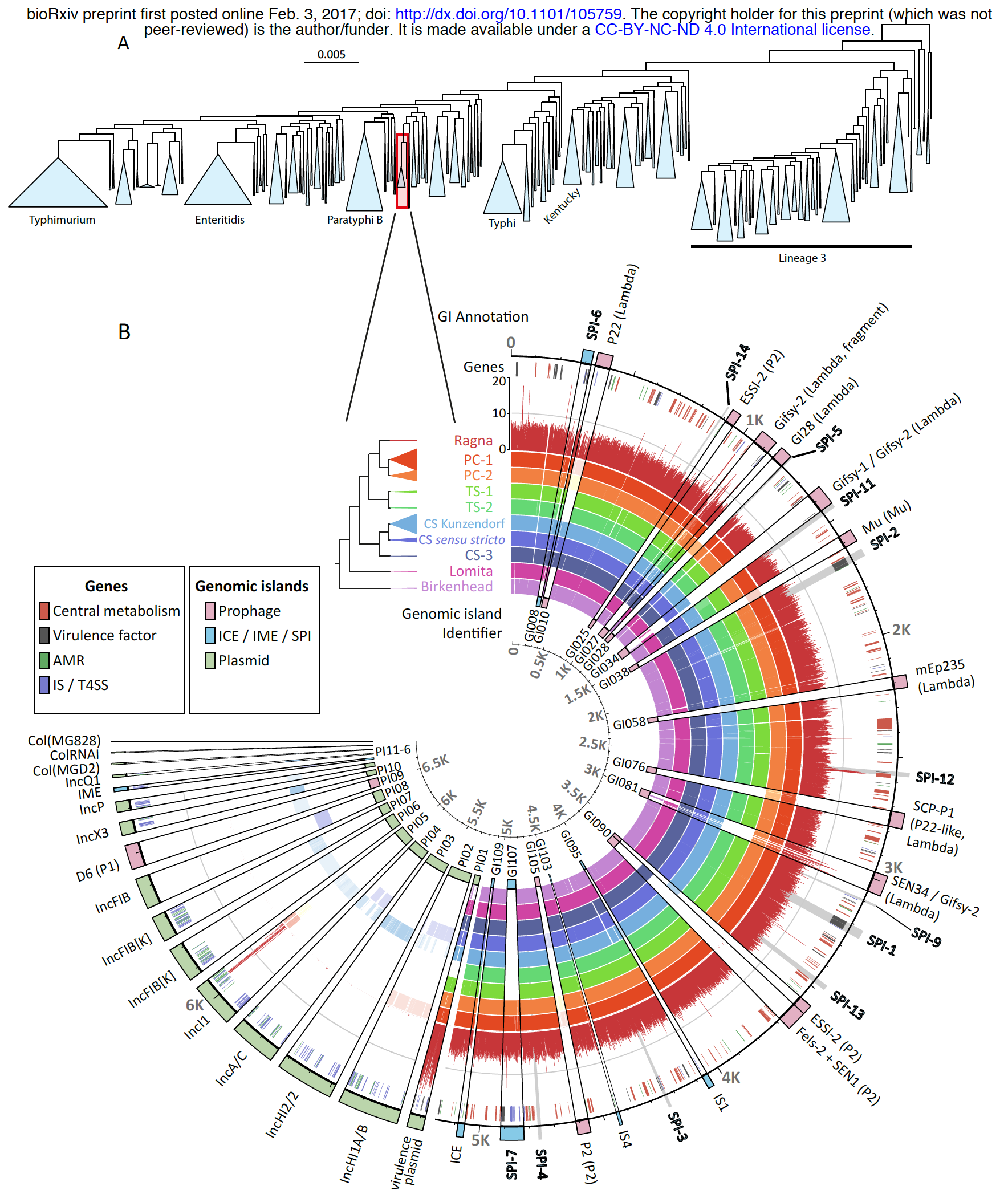
Nabil-Fareed Alikhan reminds me in the comments section under this post that they also have an interactive version of this figure on their own deployment of anvi’server under Enterobase: https://enterobase.warwick.ac.uk/anvio/public/zhemin/ParaC_pangenome.
The figure at the top shows the maximum-likelihood phylogeny of 50,000+ S. enterica genomes in the Enterobase, and at the bottom it shows the pangenome of Para C Lineage and mapping statistics of reads that likely belong to the Ragna genome. The story is already very interesting, but I can’t take my eyes from the figure.
Maybe you are familiar with the types of figures our lab generates with anvi’o. Here is an example from our paper in which we re-analyzed the contaminated Tardigrade assembly:
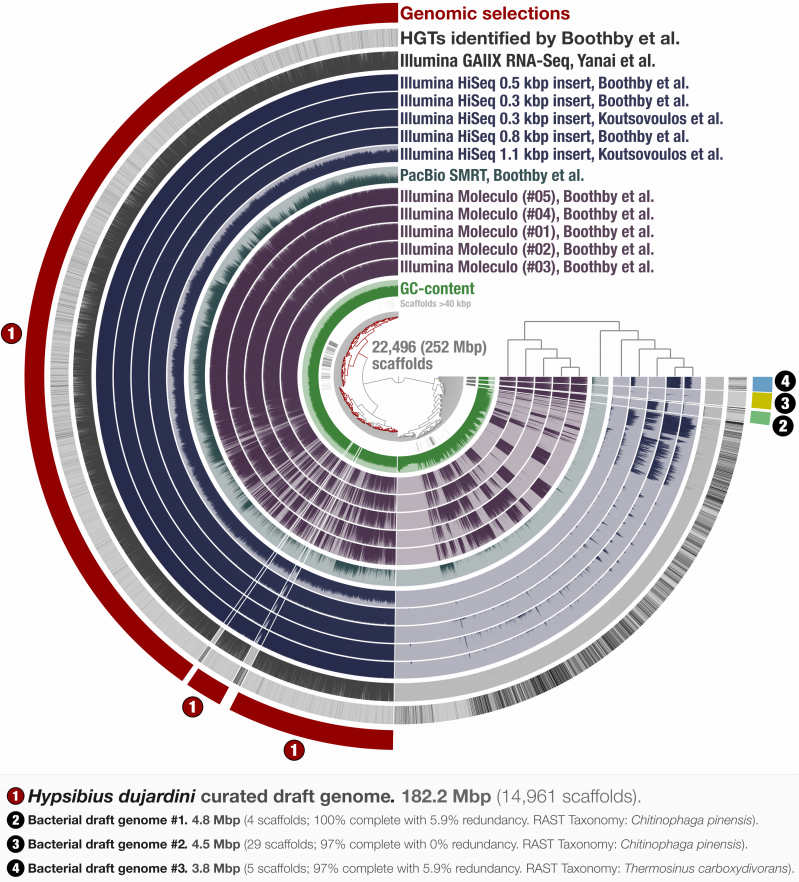
Regardless of the functionality of these figures, you probably can feel the sudden drop in aesthetics when you stop looking at Zhemin’s figure and start looking at ours.
But this is exactly what we were hoping for! From the visualization strategy to the analytical approaches we keep building into the platform, we wanted anvi’o to be free from boilerplate analysis practices. We hoped that its flexibility would allow people to use it in ways and for things we haven’t envisioned. This figure is a great example. I am looking at it, and although I know every corner of the codebase, I have no idea where would I start to generate this figure .. which makes me feel both jealous, and excited.
Every time I give a workshop, I tell people (and hear back from them) that anvi’o does have a somewhat steep learning curve. It is true. But once someone is familiar with it, anvi’o is as good in their hands as their understanding of their own research and data.
A pre-print from Mark Achtman, M. Thomas P. Gilbert, Zhemin Zhou et al, and my 2 cents on it: https://t.co/CY7Ppp8YW3
— A. Murat Eren (@merenbey) February 5, 2017