
Table of Contents
We just started playing with our human gut fecal samples using MinION to get ourselves oriented with Oxford Nanopore’s little device.
Besides all the amazing things long-read sequencers offer, they also come with multiple challenges both at the bench side (such as the difficulties associated with recovering high-molecular weight and high-concentration DNA from samples) and the computational side (such as high rates of substitution errors as well as in/dels that were out of our lives thanks to Illumina). These are particularly serious issues if your focus is genome-resolved metagenomics. Nothing in life seems to just come only with good things. #SAD.
There is not much on our end to talk about yet. Yet with this post we will start simple, and maybe we will have a series of tutorials regarding how to play with MinION data in anvi’o in the long run hopefully with your suggestions and/or questions. Do you have a clear idea what anvi’o could do for you to make sense of your MinION data? Let us know! Also feel free to drop a line if you are curious about the run time, input DNA concentration, size selection, etc.

Basically, yesterday Andrea shared with me our very first MinION results from the sequencing of one of the donor gut fecal samples from this study:
And here is a summary of what I did with it.
Creating an anvi’o contigs database
Creating a contigs database is a standard step anytime we have a bunch of reference sequences; whether these reference sequences represent contigs from an assembly, or belong to a single genome. Why should MinION output should be any different? But it is different because it comes as a FASTQ file and anvi’o works only with FASTA files.
So the first step is to convert my minion_D3.fastq (as Andrea named it) into a FASTA file using the illumina-utils library:
iu-fastq-to-fasta minion_D3.fastq \
-o minion_D3.fa
A quick note: running anvi-script-FASTA-to-contigs-db minion_D3.fa could generate an anvi’o contigs database directly from this FASTA file, but it is much better to be explicit for a learning opportunity, hence the steps below.
From this file we can create an anvi’o contigs database in multiple steps. First, we would like to simplify the deflines, so they do not contain crazy characters or long long names. This step somewhat standardizes things to decrease the likelihood of spontaneous future headaches:
anvi-script-reformat-fasta minion_D3.fa \
--simplify-names \
-o minion_D3_fixy.fa
mv minion_D3_fixy.fa minion_D3.fa
Here one could remove very short contigs from the file using the flag --min-len, but I decided to keep everything for now.
Now we have the ‘proper’ FASTA file, we can generate an anvi’o contigs database from it (during which genes would be called using prodigal, and other basic stats would be computed):
anvi-gen-contigs-database -f minion_D3.fa \
-o minion_D3.db
Finally we can run the default set of HMMs (which include profiles for ribosomal RNAs) that distribute with anvi’o on the contigs database:
anvi-run-hmms -c minion_D3.db \
--num-threads 10
Our contigs database is ready.
Taking a look at contig stats
Since the v3 release, anvi’o has a program called anvi-display-contigs-stats that visualizes basic stats of ‘things’ in a given list of contigs databases, so we can take a quick look at this database by running:
anvi-display-contigs-stats minion_D3.db
This command will open an interactive display:
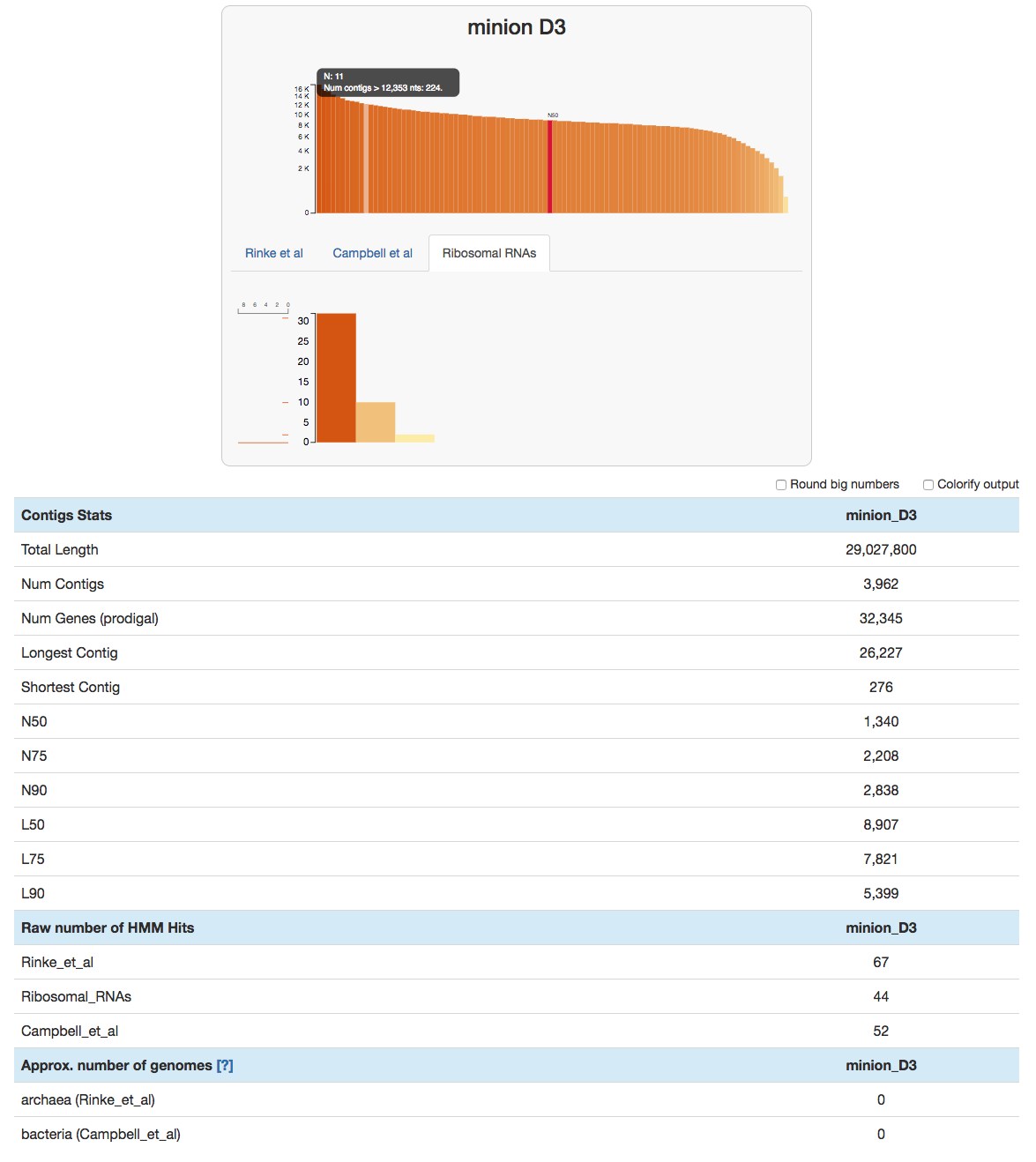
anvi-display-contigs-stats can also produce a text output, or compare multiple contigs databases (here is a screenshot for that).
{kind=link}
Most contigs are pretty well sized. Andrea’s size selection seems to have worked well.
Usually the number of rRNA genes is a fraction of our assembled metagenomes. But in this case there are as many single-copy core genes as there are rRNA hits as shown under the ‘Raw number of HMM Hits’ section. This is due to the fact that HMMs to identify ribosomal RNAs use the DNA alphabet, while the HMMs that identify single-copy core genes rely on amino acid seqeunces. Because indels cause many frame-shift errors in the gene context, amino acid sequences of those crippled gene calls become unrecognizeable to any model. DNA models continue to rock because long hair don’t care.
One of our goals going forward with MinION is to get better at correcting those errors when short read data is available. But that’s for later.
Getting out ribosomal RNA gene sequences
Since we run HMMs for ribosomal RNAs, the contigs database knows which sequences had a significant match to any of the models in our ribosomal RNA collection. We can quickly learn what models available in any HMM collection this way:
anvi-get-sequences-for-hmm-hits -c minion_D3.db \
--hmm-source Ribosomal_RNAs \
--list-available-gene-names
* Archaeal_23S_rRNA, Archaeal_5S_rRNA, Archaeal_5_8S_rRNA,
Bacterial_16S_rRNA, Bacterial_23S_rRNA, Bacterial_5S_rRNA,
Eukaryotic_28S_rRNA, Eukaryotic_5S_rRNA, Eukaryotic_5_8S_rRNA,
Mitochondrial_12S_rRNA, Mitochondrial_16S_rRNA, Archaeal_16S_rRNA
Here I want to take a quick look at 16S and 23S rRNA genes, but one could select anything from the list of available genes shown above (if you do not specify any gene names, you would get a FASTA file with everything):
anvi-get-sequences-for-hmm-hits -c minion_D3.db \
--hmm-source Ribosomal_RNAs \
--gene-name Bacterial_16S_rRNA,Bacterial_23S_rRNA \
-o minion_D3_rRNA.fa
This gives me a FASTA file with 42 sequences in it.
Naturally, I go to the NCBI with it, and BLAST them all against the nr database.
Here is the summary of top hits this script generated from the search results:
| Found in the assembly | Best hit on NCBI | Percent alignment | Percent identity | Accession |
|---|---|---|---|---|
| Bacterial_23S_rRNA | Alistipes finegoldii | 100% | 88.17% | NR_103154 |
| Bacterial_23S_rRNA | Anaerostipes hadrus | 94.82% | 81.49% | CP012098 |
| Bacterial_16S_rRNA | Bacteroides cellulosilyticus | 97.66% | 91.43% | CP012801 |
| Bacterial_23S_rRNA | Bacteroides cellulosilyticus | 94.68% | 87.26% | CP012801 |
| Bacterial_23S_rRNA | Bacteroides cellulosilyticus | 95.96% | 88.25% | CP012801 |
| Bacterial_23S_rRNA | Bacteroides ovatus | 96.61% | 87.82% | CP012938 |
| Bacterial_16S_rRNA | Bacteroides uniformis | 96.16% | 87.35% | LT745889 |
| Bacterial_23S_rRNA | Bacteroides uniformis | 94.24% | 86.61% | LT745890 |
| Bacterial_23S_rRNA | Bacteroides uniformis | 95.33% | 86.93% | LT745890 |
| Bacterial_23S_rRNA | Bacteroides uniformis | 96.14% | 84.60% | LT745890 |
| Bacterial_23S_rRNA | Bacteroides uniformis | 96.15% | 89.04% | LT745890 |
| Bacterial_23S_rRNA | Bacteroides vulgatus | 94.09% | 83.52% | CP000139 |
| Bacterial_23S_rRNA | Bacteroides vulgatus | 94.81% | 86.89% | CP000139 |
| Bacterial_23S_rRNA | Bifidobacterium longum | 95.74% | 83.44% | CP016019 |
| Bacterial_23S_rRNA | Blautia hansenii | 94.38% | 80.77% | CP022413 |
| Bacterial_16S_rRNA | Blautia luti | 96.49% | 85.06% | LC010692 |
| Bacterial_23S_rRNA | Blautia sp. | 94.10% | 79.37% | CP015405 |
| Bacterial_23S_rRNA | Burkholderiales bacterium | 94.75% | 82.78% | CP015403 |
| Bacterial_23S_rRNA | Burkholderiales bacterium | 97.68% | 81.00% | CP015403 |
| Bacterial_23S_rRNA | Clostridium sp. | 95.22% | 82.76% | FJ625862 |
| Bacterial_16S_rRNA | Dorea longicatena | 96.68% | 85.33% | LT223662 |
| Bacterial_16S_rRNA | Dorea longicatena | 97.41% | 88.00% | LC037228 |
| Bacterial_16S_rRNA | Eubacterium rectale | 95.79% | 88.58% | CP001107 |
| Bacterial_23S_rRNA | Eubacterium rectale | 92.76% | 80.10% | FP929042 |
| Bacterial_23S_rRNA | Eubacterium rectale | 94.77% | 88.93% | FP929043 |
| Bacterial_23S_rRNA | Eubacterium rectale | 96.12% | 82.80% | FP929043 |
| Bacterial_23S_rRNA | Eubacterium sp. | 94.85% | 81.55% | LT635479 |
| Bacterial_16S_rRNA | Faecalibacterium prausnitzii | 93.91% | 89.31% | CP022479 |
| Bacterial_23S_rRNA | Faecalibacterium prausnitzii | 94.37% | 85.96% | CP022479 |
| Bacterial_23S_rRNA | Faecalibacterium prausnitzii | 95.16% | 88.90% | CP022479 |
| Bacterial_23S_rRNA | Faecalibacterium prausnitzii | 96.76% | 82.23% | FP929046 |
| Bacterial_23S_rRNA | Faecalibacterium prausnitzii | 98.97% | 90.75% | FP929046 |
| Bacterial_16S_rRNA | Lachnospiraceae bacterium | 96.81% | 85.00% | LN907763 |
| Bacterial_23S_rRNA | Parabacteroides distasonis | 94.52% | 84.91% | NR_076385 |
| Bacterial_23S_rRNA | Parabacteroides distasonis | 94.91% | 82.44% | CP000140 |
| Bacterial_23S_rRNA | Ruminococcus torques | 94.80% | 84.14% | FP929055 |
| Bacterial_16S_rRNA | uncultured bacterium | 97.60% | 89.13% | EF404264 |
| Bacterial_16S_rRNA | uncultured bacterium | 98.12% | 90.72% | EF400825 |
| Bacterial_23S_rRNA | uncultured bacterium | 100.00% | 86.63% | KX127935 |
| Bacterial_23S_rRNA | uncultured bacterium | 96.00% | 77.53% | KX126288 |
| Bacterial_23S_rRNA | uncultured bacterium | 96.51% | 80.03% | KX126288 |
| Bacterial_23S_rRNA | uncultured bacterium | 99.94% | 81.09% | KX126288 |
Well.

There are some hits that resolve to the same ‘species’ name and very likely represent different operons of the same population, and others that resolve to the same ‘species’ name but closely matching to different references. Clearly abundance plays a critical role here and the number of copies of rRNA genes coming from the same population should have a linear correlation with its abundance in the metagenome. It is funny to think about the fact that we used to get almost none of them with short assembly, now with long-read sequencing we will probably get way too many.
These will get much clearer when we are done with mapping, and start looking at them in the context of the population genomes we generated using the Illumina short reads from the same donor gut.
New blog post on how to take a quick look at #MinION contigs and recover ribosomal RNA sequences from a run.
— A. Murat Eren (@merenbey) January 18, 2018
We probably will SPAM you with little bits an pieces of information like this as we start experimenting with nanopore sequencing: https://t.co/KhZc0gPUbg #anvio