

Table of Contents
The purpose of this blog post is to plant a seed in our communal mind in an attempt to encourage an open discussion. Please consider sharing your thoughts or experience with dereplication in the context of genome-resolved metagenomics. Do you dereplicate your metagenome-assembled genomes (MAGs)? Why or why not? Are you satisfied or not? What would be the ideal way to do it?
Our recent paper on bioRxiv communicates our perspective on dereplication, a computational step that is often considered when generating and analyzing genomic datasets. In theory, dereplication identifies groups of identical or highly similar genomes in a given dataset based on a user-provided sequence identity cutoff, and reduces them to a single representative genome. Although in our study we focused on MAGs, dereplication is a procedure that can be applied to sets of genomes from any source. The reason why multiple highly similar genomes are present in most MAG collections is that most studies perform single sample assemblies on a collection of samples, rather than co-assembling all data together. This has a variety of potential benefits, including the fact that multiple related populations present across samples can make assemblies more fragmented when attempting a co-assembly.
Our urge to write this perspective emerged from two observations. First, in our own work, we noticed that when performing dereplication using widely used tools, we ended up having groups of very closely related genomes, with > 99% average nucleotide identity, not being reduced to a single representative, despite using 99% identity cutoffs. Second, we started seeing multiple large collections of MAGs being released, sometimes with no dereplication step included at all.
This prompted questions as to what it means to present a certain number of genomes. Does each genome contribute to knowledge? How many of the genomes that are clustered together are actually identical to each other? How does redundancy complicate downstream analyses? On the other hand, what is lost when you do remove genomes from your dataset? Are two MAGs generated from independent metagenomic datasets ever identical? How valuable is it to repeatedly sample, at the genomic level, the same or similar populations? How do the different algorithms and their parameters affect what representatives remain? And how does dereplicating or not dereplicating your data, and genomic databases more general, bias the biological interpretations we make from our data?
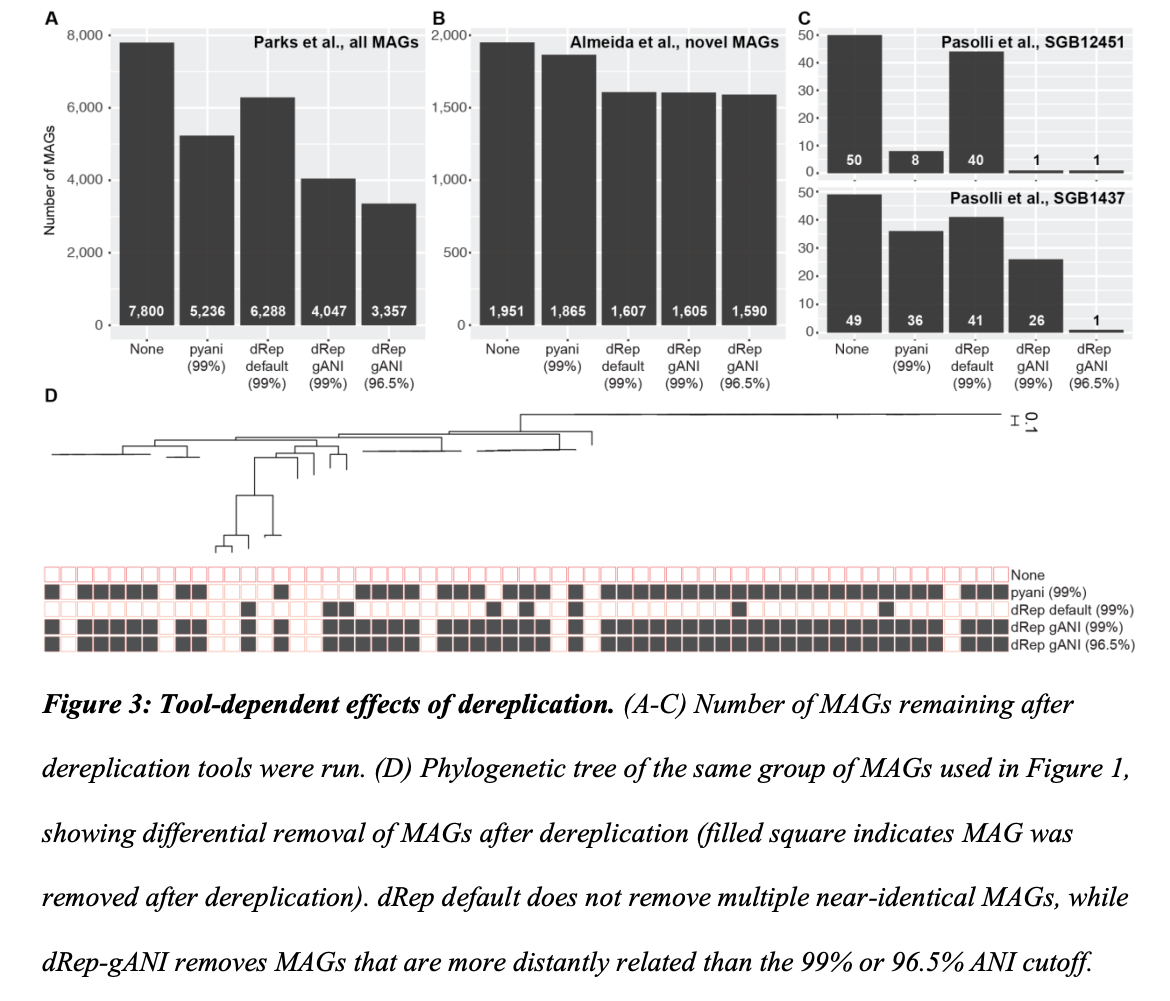
We argue that while obtaining multiple genomic representatives for a species can be highly valuable, having multiple copies of the same or highly similar genomes complicates downstream analysis. The main complication is that when redundancy in a database of genomes is maintained, the subsequent step of mapping sequencing reads back to this database of genomes leads to sequencing reads having multiple high quality alignments. When using these data to make inferences about the relative abundance and population dynamics across samples, the relative abundance for a taxon will look artificially low, and it will appear that multiple ecologically equivalent populations co-occur. While multiple closely related populations have indeed been shown to co-occur, disentangling technical issues and true biological phenomena is complicated by database redundancy combined with methods that are not taking this redundancy into account explicitly.
We also present a case study showing a risk of removing closely related genomes, specifically how this can affect the loss of information on auxiliary genes present within and between populations. While not addressed in the perspective article, loss of information on single nucleotide variants of core genes can be relevant too, although read mapping to the retained representative can reveal single nucleotide variants within and across samples. Across our analyses, we also show how commonly used tools lead to very different results, even when using the same assumed percent identity cutoff.
While our perspective only begins to answer the questions that we listed in this post, it does aim to highlight that the question whether one should perform dereplication or not, and if yes, how should they do it, requires a lot more attention and scrutiny than what many of us are giving to it. We hope it can contribute to ongoing and stimulate new discussions as to how we best move forward in our own individual studies as well as how we manage the ever increasing numbers of MAGs.
We believe the wealth of information emerging from genome-resolved metagenomics require the development of new approaches that can offer effective means to store and mine these data, and we encourage the community to continue investigating optimal strategies to get there.