Phylogeography of ribosomal proteins with EcoPhylo

Table of Contents
- Quantitative insights into the efficacy of genome-resolved surveys of microbial communities through ribosomal protein phylogeography and EcoPhylo
- Reproducible / Reusable Data Products
- Set up environment
- Distribution HMM alignment coverage and SCG detection across GTDB
- Explore SCG HMM copy number across GTDB r95 RefSeq
- Benchmarking EcoPhylo workflow with Ribosomal proteins using CAMI synthetic metagenomes
- Removing genomes that are not detected in metagenomic samples
- Identifying the most frequent ribosomal proteins in a genomic collection
- Make genome-type and miscellaneous data
- Calculating genome recovery rate
- Manually curate EcoPhylo tree
- EcoPhylo interactive interfaces
- Contemporary genome collections massively under-represent microbial diversity in marine systems
- Reproducible / Reusable Data Products
- Set up environment
- Selecting ribosomal proteins
- EcoPhylo interactive interface of the open surface ocean
- Scatter plot of the number of SCGs recovered in assembled metagenomes vs sequencing depth from 237 surface ocean metagenomes.
- Regression analysis of latitude versus bacterial + archaeal rpL14 richness from surface ocean metagenomes
- Log mean Q2Q3 coverage of bacteria and archaea rpL14 sequences by genome type across surface ocean.
The purpose of this page is to provide access to reproducible data products that underlie our key findings from two studies by Matt Schechter et al. including: “Quantitative insights into the efficacy of genome-resolved surveys of microbial communities through ribosomal protein phylogeography and EcoPhylo” and “Contemporary genome collections massively under-represent microbial diversity in marine systems”.
This reproducible workflow begins with “Quantitative insights into the efficacy of genome-resolved surveys of microbial communities through ribosomal protein phylogeography and EcoPhylo” which introduces the development and benchmarking of the anvi’o EcoPhylo workflow using ribosomal proteins to evaluate genome recovery from microbial communities. The latter half of the document will document the reproducbile workflow for “Contemporary genome collections massively under-represent microbial diversity in marine systems” which applies the EcoPhylo workflow and ribosomal proteins to the global surface ocean. If you would like to skip directly to “Contemporary genome collections massively under-represent microbial diversity in marine systems” then click here.
If you have any questions and/or if you are unable to find an important piece of information here, please feel free to leave a comment down below, send an e-mail to us, or get in touch with us through Discord:

Quantitative insights into the efficacy of genome-resolved surveys of microbial communities through ribosomal protein phylogeography and EcoPhylo
- In which we show the application of EcoPhylo to ribosomal proteins to be able to investigate the genome recovery rates from metagenomes and demonstrate its efficacy across three biomes using genomes and metagenomes from the human gut, human oral cavity, and surface ocean.
- A reproducible bioinformatics workflow for the study is availabe here.
Reproducible / Reusable Data Products
The following data items are compatible with anvi’o version v8 or later. The anvi’o contigs-db and profile-db in them can be further analyzed using any program in the anvi’o ecosystem, or they can be used to report summary data in flat-text files to be imported into other analysis environments.
- DOI:10.6084/m9.figshare.28207481.v2: Ribosomal protein interative interfaces for the human oral cavity (rpL19, rpS15, rpS2) and human gut (rpS16, rpS19, rpS15). To directly load this data, skip to the section EcoPhylo interactive interfaces
Set up environment
If you want to follow along and run each code block in the following reproducible, you will need to install anvi’o. Next you will need to download the associated data (DOI:10.6084/m9.figshare.28208018) and open it up like this:
curl -L -o REPRODUCIBLE_WORKFLOW_DATA.tar.gz https://api.figshare.com/v2/file/download/51683177
tar -xvzf REPRODUCIBLE_WORKFLOW_DATA.tar.gz
Load packages
These are the r packages you will need to load and install before running the analysis:
packages <- c("tidyverse", "ggpubr", "fs", "ape", "treeio", "glue", "plotly", "readxl")
suppressWarnings(suppressMessages(lapply(packages, library, character.only = TRUE)))
The majority of commands using anvi-run-workflow were run on high performance compute clusters (HPC) leveraging the powerful SLURM wrapper clusterize. Without access to compute nodes, these commands would take a VERY long time. Please check out this blogpost for more information: Running workflows on a cluster. All BASH commands in this reproducible workflow will need to be modified to run on your own HPC system. See below for an example some modifiable paramaters that might be helpful.
Here is an “example” of prototypical command with some basic descriptions of the various parameters. Make sure to fill in everything on your own :)
anvi-run-workflow -w ecophylo \ # anvio worklow name
-c config.json \ # workflow config file
--additional-params \ # extract parameters that go straight to snakemake
--cluster "qsub {threads}" \ # example string to send jobs to HPC
--jobs=120 \ # number of co-occuring jobs
--resources nodes=120 \ # num. of nodes the workflow can use
--latency-wait 100 \ # wait 100 sec. for output file to appear
--keep-going \ # finish as many jobs as possible even if one job fails
--rerun-incomplete # restart any jobs that failed
Distribution HMM alignment coverage and SCG detection across GTDB
To identify a threshold to remove spurious hmm-hits recruited by ribosomal protein HMMs in metagenomic assemblies, we examined the distribution of hmm-hits alignment coverage across genomes across GTDB release 95 and decided to filter our hmm-hits with less than 80% model coverage. The idea here was to improve our detection of high quality hmm-hits in genomes in an effort to improve the detection of ribosomal proteins in the wild, metagenomic assemblies. The following steps describe our workflow:
Step 1. Run EcoPhylo over GTDB Bacteria and Archaea collections with the Bacteria_71 and Archaea_76 HMM collections respectively with NO HMM alignment coverage cutoff.
To run this, you will need to make external-genomes with paths to all contigs-db for GTDB genomes.
# get default config
anvi-run-workflow -w ecophylo --get-default-config ecophylo_config_GTDB_reps.json
# Make hmm_list.txt
echo -e "name\tsource\tpath" > hmm_list.txt
echo -e "Ribosomal_L14\tBacteria_71\tINTERNAL" >> hmm_list.txt
# Make log dir
mkdir -p ECOPHYLO_WORKFLOW_GTDB_REPS/00_LOGS
# Run workflow
anvi-run-workflow -w ecophylo \
-c ecophylo_config_GTDB_reps.json \
--skip-dry-run \
--additional-params \
--latency-wait 100 \
--keep-going \
--rerun-incomplete
Here is how you can adjust the config.json to not filter based on HMM
alignment coverage:
{
"filter_hmm_hits_by_model_coverage": {
"threads": 1,
"--model-coverage": 0.001,
"additional_params": ""
},
}
Step 2. Extract hmmsearch domtblout files
Here we iterated through all of the domtblout files that were produced and concatenated them. They can be found in an EcoPhylo dir structure here: 01_REFERENCE_PROTEIN_DATA/*/*-dom-hmmsearch/hmm.domtable
PROTEIN="" # replace with protein interest
for file in `ls ECOPHYLO_WORKFLOW/01_REFERENCE_PROTEIN_DATA/*/"${PROTEIN}"-dom-hmmsearch/hmm.domtable`;
do
fname=$(dirname "${file}" | sed 's|/"${PROTEIN}"-dom-hmmsearch||' | sed 's|ECOPHYLO_WORKFLOW/01_REFERENCE_PROTEIN_DATA/||')
echo -e "${fname}"
sed "s/^/"${fname}"\t&/g" "${file}" >> hmm.domtable.GTDB.txt;
done
Step 3. Clean the domtblout data
Here we imported the concatenated hmm.domtable tables we made above
and cleaned up the data to plot.
DIR_PATH <- "REPRODUCIBLE_WORKFLOW_DATA"
# Read in domtlout files
GTDB_domtable <- read_table(path(DIR_PATH, "hmm.domtable.GTDB.txt"), col_names = FALSE)
GTDB_domtable_archaea <- read_table(path(DIR_PATH, "hmm.domtable.GTDB.archaea.txt"), col_names = FALSE)
fix_domtblout_data <- function(X) {
X %>%
rename("genome" = X1,
"gene_callers_id" = X2,
"target_accession" = X3,
"gene_length" = X4,
"hmm_name" = X5,
"hmm_id" = X6,
"hmm_length" = X7,
"evalue" = X8,
"bitscore" = X9,
"bias" = X10,
"match_num" = X11,
"num_matches" = X12,
"dom_c_evalue" = X13,
"dom_i_evalue" = X14,
"dom_bitscore" = X15,
"dom_bias" = X16,
"hmm_start" = X17,
"hmm_stop" = X18,
"gene_start" = X19,
"gene_stop" = X20,
"env_to" = X21,
"env_from" = X22,
"mean_post_prob" = X23,
"description" = X24) %>%
mutate(model_coverage = (hmm_stop - hmm_start)/hmm_length) %>%
mutate(gene_coverage = (gene_stop - gene_start)/gene_length) %>%
select(genome, gene_callers_id, hmm_name, model_coverage, gene_coverage) %>%
arrange(desc(model_coverage))
}
GTDB_domtable_ed <- fix_domtblout_data(GTDB_domtable)
GTDB_domtable_archaea_ed <- fix_domtblout_data(GTDB_domtable_archaea)
Step 4. Plot the distribution of model and gene alignment coverages
This is how we plotted the distribution of HMM alignment coverages to all the ORF hits across the genome data sets. The files GTDB_domtable_ed.tsv and GTDB_domtable_archaea_ed.tsv and saved in the datapack.
DIR_PATH <- "REPRODUCIBLE_WORKFLOW_DATA"
GTDB_domtable_ed <- read_tsv(path(DIR_PATH, "GTDB_domtable_ed.tsv"))
GTDB_domtable_archaea_ed <- read_tsv(path(DIR_PATH, "GTDB_domtable_archaea_ed.tsv"))
plot_boxplot_HMM_alignment_coverage_distribution <- function(X, HMM_source) {
plot_model_cov <- X %>%
group_by(hmm_name) %>%
mutate(avg = mean(model_coverage),
median = median(model_coverage)) %>%
ggplot(aes(x=model_coverage, y=fct_reorder(hmm_name, median))) +
geom_boxplot(outlier.size = 0.5,
outlier.shape = 1,
outlier.alpha = 0.5) +
geom_vline(xintercept = 0.8, color = "blue") +
geom_vline(xintercept = 0.95, color = "red") +
theme_light() +
theme(legend.position = "none",
axis.ticks.x=element_blank(),
legend.title = element_blank(),
axis.text=element_text(size = 6),
text=element_text(family="Helvetica Neue Condensed Bold"),
plot.title = element_text(hjust = 0.5)
) +
xlab("HMM model alignment coverage") +
ylab(str_c(HMM_source, "model ordered by mean coverage", sep = " ")) +
ggtitle(str_c("hmmsearch model coverage for", HMM_source, sep = " "))
plot_gene_cov <- X %>%
group_by(hmm_name) %>%
mutate(avg = mean(gene_coverage),
median = median(gene_coverage)) %>%
ggplot(aes(x=gene_coverage, y=fct_reorder(hmm_name, median))) +
geom_boxplot(outlier.size = 0.5,
outlier.shape = 1,
outlier.alpha = 0.5) +
geom_vline(xintercept = 0.8, color = "blue") +
geom_vline(xintercept = 0.95, color = "red") +
theme_light() +
theme(legend.position = "none",
axis.ticks.x=element_blank(),
legend.title = element_blank(),
axis.text=element_text(size = 6),
text=element_text(family="Helvetica Neue Condensed Bold"),
plot.title = element_text(hjust = 0.5)
) +
xlab("gene alignment coverage") +
ylab(str_c(HMM_source, "gene ordered by mean coverage", sep = " ")) +
ggtitle(str_c("hmmsearch gene-coverage for", HMM_source, sep = " "))
list(plot_model_cov = plot_model_cov,
plot_gene_cov = plot_gene_cov)
}
plot_model_cov_bacteria_71 <- plot_boxplot_HMM_alignment_coverage_distribution(GTDB_domtable_ed,
HMM_source = "Bacteria_71")
plot_model_cov_archaea_76 <- plot_boxplot_HMM_alignment_coverage_distribution(GTDB_domtable_archaea_ed,
HMM_source = "Archaea_76")
ggarrange(plot_model_cov_archaea_76$plot_model_cov,
plot_model_cov_bacteria_71$plot_model_cov,
plot_model_cov_archaea_76$plot_gene_cov,
plot_model_cov_bacteria_71$plot_gene_cov,
ncol = 2,
nrow = 2)

Explore SCG HMM copy number across GTDB r95 RefSeq
To identify ribosomal proteins that occurred in single-copy per genome, we searched the Bacteria_71 and Archaea_76 HMM collections across GTDB with 80% model alignment coverage (threshold identified in the previous analysis to filter out low quality HMM-hits in metagenomic assembles).
Step 1. Run EcoPhylo with 80% HMM alignment coverage cut-off
# Bacteria
# get default config
anvi-run-workflow -w ecophylo --get-default-config ecophylo_config_GTDB_reps.json
# Make log dir
mkdir -p ECOPHYLO_WORKFLOW_GTDB_REPS_80/00_LOGS
# Run workflow
anvi-run-workflow -w ecophylo \
-c ecophylo_config_GTDB_reps_80.json \
--skip-dry-run \
--additional-params \
--latency-wait 100 \
--keep-going \
--rerun-incomplete
Here is how you can adjust the config.json to filter for 80% HMM alignment coverage:
{
"filter_hmm_hits_by_model_coverage": {
"threads": 1,
"--model-coverage": 0.8,
"additional_params": ""
},
}
Step 2. Extract matrix of hmm-hits across genome data sets
This is how we extracted a matrix of hmm-hits for the HMM collections (Bacteria_71 and Archaea_76) after running the EcoPhylo workflow
over the GTDB representative genomes dataset. The script anvi-script-gen-hmm-hits-matrix-across-genomes is a convenient tool to extract an hmm-hits count matrix from any set of genomes and/or metagenomes in anvi’o.
# archaea
anvi-script-gen-hmm-hits-matrix-across-genomes -e external-genomes-archaea.txt \
--hmm-source Archaea_76 \
-o GTDB_representatives_refseq_external_genomes_archaea_GENOME_MATRIX.txt
# bacteria
anvi-script-gen-hmm-hits-matrix-across-genomes -e external-genomes-archaea.txt \
--hmm-source Bacteria_71 \
-o GTDB_representatives_refseq_external_genomes_bacteria_GENOME_MATRIX.txt
Step 3. Count copy number of SCGs across genomes
DIR_PATH <- "REPRODUCIBLE_WORKFLOW_DATA"
# load data
SCG_MATRIX_ARCHAEA <- read_tsv(path(DIR_PATH, "GTDB_representatives_refseq_external_genomes_archaea_GENOME_MATRIX.txt"))
SCG_MATRIX_BACTERIA <- read_tsv(path(DIR_PATH, "GTDB_representatives_refseq_external_genomes_bacteria_GENOME_MATRIX.txt"))
# make ggplot theme to beautify the figure
plot_theme <- theme_light() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1, family = "Helvetica Neue Condensed Bold"),
axis.ticks.x = element_blank(),
legend.title = element_blank(),
axis.text = element_text(size = 12, family = "Helvetica Neue Condensed Bold"),
axis.title = element_text(size = 18, family = "Helvetica Neue Condensed Bold"),
text = element_text(size = 8, family = "Helvetica Neue Condensed Bold"),
plot.title = element_text(hjust = 0.5, size = 18, family = "Helvetica Neue Condensed Bold"),
legend.position = "bottom")
# This function will plot the proportions copy number across all genomes for each SCG HMM
plot_num_scg <- function(X, TITLE, XAXIS, YAXIS) {
SCG_MATRIX_long <- X %>% pivot_longer(-genome_or_bin) %>% rename(SCG = "name")
# Get order of bar charts
percent_order <- X %>%
pivot_longer(-genome_or_bin) %>%
rename(SCG = "name") %>%
mutate(presence = case_when(value >= 1 ~ 1,
value < 1 ~ 0)) %>%
group_by(SCG) %>%
summarize(total = sum(presence)) %>%
mutate(percent = (total/SCG_MATRIX_long$genome_or_bin %>% unique() %>% length())*100)
num_scgs <- SCG_MATRIX_long %>% .$SCG %>% unique() %>% length()
num_genomes <- SCG_MATRIX_long %>% .$genome_or_bin %>% unique() %>% length()
SCG_MATRIX_long %>%
group_by(SCG) %>%
count(value) %>%
mutate(value = as.character(value),
once = n[value == "1"]) %>%
left_join(percent_order) %>%
ggplot(aes(x = fct_reorder(SCG, percent), y = n/num_genomes, fill = value)) +
geom_bar(stat="identity") +
plot_theme +
ylab(XAXIS) +
xlab(YAXIS) +
ggtitle(glue(TITLE, " (n = {num_genomes} genomes)"))
}
SCG_count_archaea <- plot_num_scg(SCG_MATRIX_ARCHAEA,
XAXIS = "Percent detection GTDB Archaea genomes",
YAXIS = "Archaea_76 SCG Pfam models",
TITLE = "Distribution of SCG hits per GTDB Archaea representative genome")
SCG_count_bacteria <- plot_num_scg(SCG_MATRIX_BACTERIA,
XAXIS = "Percent detection GTDB Bacteria genomes",
YAXIS = "Bacteria_71 SCG Pfam models",
TITLE = "Distribution of SCG hits per GTDB Bacteria representative genome")
plot_final <- ggarrange(SCG_count_bacteria,
SCG_count_archaea,
nrow = 2,
labels = c("A", "B"), font.label = (family = "Helvetica Neue Condensed Bold")) %>%
annotate_figure(top = text_grob("SCG HMM detection across GTDB r95 representative genomes", size = 24, family = "Helvetica Neue Condensed Bold"))
plot_final

Benchmarking EcoPhylo workflow with Ribosomal proteins using CAMI synthetic metagenomes
We explored different ribosomal protein clustering thresholds and their impact on non-specific read recruitment in the EcoPhylo workflow. To do this, we used CAMI and performed a grid search across ribosomal protein clustering thresholds from 95%-100%. Before we started, we identified the top 5 most frequent single-copy ribosomal proteins per CAMI genome dataset. Here is an example of how we performed the read recruitment experiment with the CAMI marine dataset.
Step 1. Make config files for ribosomal protein clustering grid search from 95%-100%
Here we used the script paramaterize_clustering_threshold.py to iterate through the parameters we were testing and modify a default
config.json accordingly.
# Get default ecophylo config file
anvi-run-workflow -w ecophylo --get-default-config ecophylo.json
# Edit config file to perform grid search
DATASET="MARINE"
for DIR_SCG in RP_L22 RP_L2 RP_L5 RP_15 RP_S8
do
for i in 95 96 97 98 99 1
do
python /SCRIPTS/paramaterize_clustering_threshold.py --input-config-json ecophylo.json \
--output-config-json ecophylo_"${DIR_SCG}"_"${i}"_"${DATASET}".json \
--min-seq-id $i \
--cov-mode 1 \
--hmm-list hmm_list_"${DIR_SCG}".txt \
--SCG "${DIR_SCG}" \
--cami-dataset "${DATASET}";
done
done
Step 2. Run EcoPhylo workflow over grid search
This is how we ran EcoPhylo over the CAMI Marine dataset with multiple ribosomal proteins and at a 95% clustering threshold. We performed this over each CAMI dataset with all clustering thresholds from 95-100%.
DATASET="MARINE"
for DIR_SCG in RP_L22 RP_L2 RP_L5 RP_15 RP_S8
do
mkdir -p ECOPHYLO_WORKFLOW_95_PERCENT_SIM_"${DIR_SCG}"_"${DATASET}"/00_LOGS
anvi-run-workflow -w ecophylo -c ecophylo_"${DIR_SCG}"_95_"${DATASET}".json --skip-dry-run --additional-params --cluster \"clusterize -j={rule} -o={log} -n={threads} --exclude \'\' -x \" --jobs=120 --resources nodes=120 --latency-wait 100 --keep-going --rerun-incomplete
done
Step 3. Collect non-specific read recruitment data
Here is how we iterated over the EcoPhylo directory structure to extract the non-specific read recruitment data.
# Collect mismapping data
MISMAPPING_FILE_NAME="mismapping_MARINE.tsv"
DATASET="MARINE"
echo -e "SCG\tclustering_threshold\tsample\tTotal_reads_mapped\tMAQ_lessthan_2\tnum_mismapping" > "$MISMAPPING_FILE_NAME"
# Loop through the desired directories and collect missmapping data
for DIR_SCG in RP_L22 RP_L2 RP_L5 RP_15 RP_S8
do
for i in 95 96 97 98 99 1
do
RP=$(echo "$DIR_SCG" | sed 's|RP_||')
MAPPING_PATH="CAMI/${DATASET}/ECOPHYLO_WORKFLOW_${i}_PERCENT_SIM_${DIR_SCG}_${DATASET}/METAGENOMICS_WORKFLOW/04_MAPPING/Ribosomal_${RP}"
# Loop through the BAM files in the directory
for sample in "$MAPPING_PATH"/*bam
do
SAMPLE_NAME=$(basename "$sample")
NUM_MISMAPPING=$(samtools view "$sample" | grep XS:i | cut -f12-13 | sed 's/..:i://g' | awk '$1==$2' | wc -l) # magic command to collect missmapping data
TOTAL_NUM_READS_MAPPED=$(samtools view "$sample" | wc -l)
MAPQ_lessthan_2=$(samtools view "$sample" | awk '$5<2' | wc -l)
echo -e "$DIR_SCG\t$i\t$SAMPLE_NAME\t$TOTAL_NUM_READS_MAPPED\t$MAPQ_lessthan_2\t$NUM_MISMAPPING" >> "$MISMAPPING_FILE_NAME"
done
done
done
Step 4. Plot the percent of non-specific read recruitment per sample
This script will plot the percent of non-specific read recruitment per protein per metagenomes in all three CAMI datasets.
plot_theme_2 <- theme_light() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1, family = "Helvetica Neue Condensed Bold"),
axis.ticks.x = element_blank(),
legend.title = element_blank(),
axis.text = element_text(size = 8, family = "Helvetica Neue Condensed Bold"),
axis.title = element_text(size = 8, family = "Helvetica Neue Condensed Bold"),
text = element_text(size = 8, family = "Helvetica Neue Condensed Bold"),
plot.title = element_text(hjust = 0.5, size = 8, family = "Helvetica Neue Condensed Bold"),
legend.position = "bottom")
plot_missmapping <- function(PATH, TITLE){
mismapping_MARINE <- read_tsv(PATH)
mismapping_MARINE_ed <- mismapping_MARINE %>%
mutate(percent_mismapping = (num_mismapping/Total_reads_mapped),
clustering_threshold = as_factor(clustering_threshold))
mismapping_MARINE_ed$clustering_threshold <- factor(mismapping_MARINE_ed$clustering_threshold, levels = c('95','96', '97', '98', '99', '1'), ordered = TRUE)
mismapping_MARINE_ed %>%
ggplot(aes(x = clustering_threshold, y = percent_mismapping, fill = SCG)) +
geom_boxplot() +
scale_y_continuous(labels = scales::percent) +
plot_theme_2 +
ggtitle(TITLE)
}
# Plot missmapping
DIR_PATH <- "REPRODUCIBLE_WORKFLOW_DATA"
MARINE_missmapping <- plot_missmapping(path(DIR_PATH, "mismapping_MARINE.tsv"), TITLE = "Marine")
STRAIN_MADNESS_missmapping <- plot_missmapping(path(DIR_PATH, "mismapping_STRAIN_MADNESS.tsv"), TITLE = "STRAIN MADNESS")
PLANT_ASSOCIATED_missmapping <- plot_missmapping(path(DIR_PATH, "mismapping_PLANT_ASSOCIATED.tsv"), TITLE = "PLANT ASSOCIATED")
missmapping_p <- ggarrange(MARINE_missmapping,
STRAIN_MADNESS_missmapping,
PLANT_ASSOCIATED_missmapping,
ncol = 3, nrow = 1,
labels = c("A", "B", "C"))
missmapping_p

Removing genomes that are not detected in metagenomic samples
When contextualizing a genomic collection from multiple genome recovery methods, some genomes will inherently be undetected in the underlying metagenomes. It’s best practice to remove these from the EcoPhylo interface so that every branch in the phylogenetic tree is relevant to the analysis and the detection heatmap doesn’t have a bunch of zeros. For example, in this manuscript we augmented Shaiber et al. (2020) MAGs with HOMD isolate genomes. Many HOMD isolate genomes were isolated from other samples and thus not detected in the Shaiber et al. (2020) metagenomes.
EcoPhylo offers an efficient way to see if genomes are detected in a metagenomes because read recruitment to ribosomal proteins is much faster than to whole genomes. We used this feature to quickly filter out genomes that are not detected in any metagenomes we analyzed. Subsequently, we re-run the workflow with the a filtered external-genomes file containing only genomes that are detected in the data. Here is an outline we did this in the paper:
Step 1. Run the EcoPhylo workflow with the entire genome collection
Step 2. Export the detection of each ribosomal protein
This program will extract a matrix of read recruitment detection data from each ribosomal protein homologue across the dataset of metagenomes using EcoPhylo:
anvi-export-table PROFILE.db --table detection_splits -o detection_splits.txt
Here is a function we used to filter for undetected genomes in the metagenomic dataset. We defined detected as 90% coverage of a ribosomal protein sequence representative in at least one metagenome. The exported list of genomes were undetected and can be used to filter out genomes from your exteral-genomes.txt.
detect_and_write_genomes <- function(DIR_PATH, SCG, DETECTION_SPLITS, DETECTION_VALUE) {
detection_splits <- read_tsv(path(DIR_PATH, DETECTION_SPLITS))
mmseqs_NR_cluster <- read_tsv(path(DIR_PATH, glue("{SCG}-mmseqs_NR_cluster.tsv")),
col_names = c("representative", "cluster_member"))
splits_0_detection <- filter_splits_by_detection(detection_splits, DETECTION_VALUE, SCG)
genomes_not_detected <- mmseqs_NR_cluster %>%
separate(representative, into = c("representative", "representative_split"), sep = glue("_{SCG}_")) %>%
separate(cluster_member, into = c("cluster_member", "cluster_member_split"), sep = glue("_{SCG}_")) %>%
select(representative, cluster_member) %>%
filter(representative %in% splits_0_detection$representative_name) %>% # Filter for splits that were not detected in any sample
select(cluster_member) %>%
filter(grepl("^GCA_", cluster_member)) # Filter for only HOMD genomes (PLEASE SEE NOTE BELOW)
}
DIR_PATH <- "PATH/TO/ECOPHYLO_DIRECTORY"
SCG <- "Ribosomal_L19" # Here you can plug in your ribosomal protein name you have in your hmm_list.txt
genomes_NOT_detected_Ribosomal_L19 <- detect_and_write_genomes(DIR_PATH, SCG, DETECTION_VALUE = 0.9)
NOTE: In this paper we employed a conservative detection statistic for ribosomal proteins in metagenomic data: 90% anvi’o detection. This could in theory filter out some ribosomal proteins that originated from the metagenomic assembly. However, we decided to keep these sequences since they were assembled from the underlying metagenomes. This line of the function is a great place to ONLY external genomes from other genome recovery method, such as isolate genomes and SAGs.
Step 3. Re-run EcoPhylo with a filtered external-genomes file with only genomes that were detected in the metagenomic dataset.
Identifying the most frequent ribosomal proteins in a genomic collection
Selecting which ribosomal protein or gene family to analyze in EcoPhylo is a critical decision that must align with your specific scientific questions. For this study, we prioritized ribosomal proteins that supported our goal of examining genome recovery rates. Specifically, we chose ribosomal proteins that (1) were present as single-copy genes across the majority of genome collections in the oral and human gut metagenomic datasets, providing broad contextualization, and (2) exhibited balanced assembly rates in metagenomic assemblies, ensuring they were neither over- nor under-assembled.
Here is an example of how we identified the top three proteins to contextualize MAGs and HOMD isolates in Shaiber et al.(2020) metagenomes.
Step 1. Run EcoPhylo workflow to annotate HOMD with Bacteria_71 SCG collection
Step 2. Find SCGs that detect the majority of the genomic dataset
Here we used anvi-script-gen-hmm-hits-matrix-across-genomes to extract the matrix of hmm-hits from both the metagenomic assemblies and genome collection.
# Exctract SCGs across metagenome dataset
anvi-script-gen-hmm-hits-matrix-across-genomes -e metagenomes.txt \
--hmm-source Bacteria_71 \
-o Bacteria_71_Oral_METAGENOME_MATRIX.txt
# Exctract SCGs genome collection dataset
anvi-script-gen-hmm-hits-matrix-across-genomes -e external_genomes_MAGs_HOMD.txt \
--hmm-source Bacteria_71 \
-o Bacteria_71_Oral_GENOME_MATRIX.txt
Step 3. Plot ribosomal protein detection across metagenomic assemblies
DIR_PATH <- "REPRODUCIBLE_WORKFLOW_DATA"
Bacteria_71_METAGENOME_MATRIX <- read_tsv(path(DIR_PATH, "Bacteria_71_Oral_METAGENOME_MATRIX.txt"))
SCG_detection_plot_theme <- theme_light() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1, family = "Helvetica Neue Condensed Bold"),
axis.ticks.x = element_blank(),
legend.title = element_blank(),
axis.text = element_text(size = 6, family = "Helvetica Neue Condensed Bold"),
axis.title = element_text(size = 6, family = "Helvetica Neue Condensed Bold"),
text = element_text(size = 6, family = "Helvetica Neue Condensed Bold"),
plot.title = element_text(hjust = 0.5, size = 8, family = "Helvetica Neue Condensed Bold"))
plot_metagenome_SCGs <- function(X, YLAB, XLAB, TITLE) {
X_long <- X %>%
pivot_longer(-genome_or_bin) %>%
rename(SCG = "name") %>%
group_by(SCG) %>%
summarize(total = sum(value))
mean_SCG_count <- X_long$total %>% mean() %>% round()
X_long %>%
ggplot(aes(x = total, y = fct_reorder(SCG, total))) +
geom_point() +
scale_x_continuous(labels=scales::comma) +
geom_vline(xintercept = mean_SCG_count, color = "blue") +
SCG_detection_plot_theme +
ylab(YLAB) +
xlab(XLAB) +
ggtitle(glue("Single-copy core gene frequencies across {TITLE} (n = {X$genome_or_bin %>% length()}; mean = {mean_SCG_count})"))
}
plot_metagenome_SCGs_frequencies <- plot_metagenome_SCGs(Bacteria_71_METAGENOME_MATRIX,
YLAB = "Single-copy core genes (anvi'o Bacteria_71)",
XLAB = "Total SCG counts",
TITLE = "oral metagenomes")
Step 4. Plot ribosomal protein detection across genome dataset
DIR_PATH <- "REPRODUCIBLE_WORKFLOW_DATA"
Bacteria_71_GENOME_MATRIX <- read_tsv(path(DIR_PATH, "Bacteria_71_Oral_GENOME_MATRIX_HOMD_MAGs.txt"))
plot_genome_SCGs <- function(X, TITLE, XLAB, YLAB) {
SCG_MATRIX_long <- X %>% pivot_longer(-genome_or_bin) %>% rename(SCG = "name")
# Get order of bar charts
percent_order <- X %>%
pivot_longer(-genome_or_bin) %>%
rename(SCG = "name") %>%
mutate(presence = case_when(value >= 1 ~ 1,
value < 1 ~ 0)) %>%
group_by(SCG) %>%
summarize(total = sum(presence)) %>%
mutate(percent = (total/SCG_MATRIX_long$genome_or_bin %>% unique() %>% length())*100)
num_scgs <- SCG_MATRIX_long %>% .$SCG %>% unique() %>% length()
num_genomes <- SCG_MATRIX_long %>% .$genome_or_bin %>% unique() %>% length()
SCG_MATRIX_long %>%
group_by(SCG) %>%
count(value) %>%
mutate(percent_occ = n/sum(n)*100) %>%
mutate(value = as.character(value),
once = n[value == "1"]) %>%
left_join(percent_order) %>%
ggplot(aes(x = fct_reorder(SCG, percent), y = percent_occ, fill = value)) +
geom_bar(stat="identity") +
SCG_detection_plot_theme +
theme(legend.position="left") +
ylab(XLAB) +
xlab(YLAB) +
ggtitle(TITLE)
}
plot_bacteria_SCG_frequency_per_HOMD_MAGs_50 <- plot_genome_SCGs(Bacteria_71_GENOME_MATRIX,
YLAB = "Single-copy core genes (anvi'o Bacteria_71)",
XLAB = "Percent of genome collection detected",
TITLE = glue("Single-copy core gene percent frequencies across oral genome dataset (HOMD + MAGs))"))
ggarrange(plot_bacteria_SCG_frequency_per_HOMD_MAGs_50, plot_metagenome_SCGs_frequencies, ncol = 2)

Make genome-type and miscellaneous data
EcoPhylo provides some basic miscellaneous information about each of the ribosomal proteins detected in your data including: representative sequence length, cluster size, and taxonomic assignment for ribosomal proteins. In this paper, we needed to add more miscellaneous data to each EcoPhylo interface, specifically the source of each ribosomal protein (MAG, SAG, isolate genome) to calculate genome recovery rates. Luckily, anvi’o makes it easy to enrich the interface with extra data to get more insights.
There are some key files you will need to create miscellaneous data for every sequence cluster in the EcoPhylo interactive interface:
-
collection-DEFAULT.txt: This file contains all the names of the splits in the EcoPhylo interactive interface. -
02_NR_FASTAS/PROTEIN-mmseqs_NR_cluster.tsv: This file contains all of the cluster representatives and cluster members. This file is critical because most of the time you want to annotate aspects of the clusters not just the representative sequences :)
Leveraging a combination of files should allow you to make miscellaneous data about any EcoPhylo sequence cluster! Here are the steps to make a miscellaneous data table with the genome-type composition for each cluster:
Step 1. Get default collection collection-DEFAULT.txt to make
metadata
anvi-export-collection -p PROFILE.db -C DEFAULT
Step 2. Create genome type metadata
Here is an example of how I made a genome-types file for the Oral Microbiome data analysis. Please note that the code is not generalizable to any miscellaneous data scenario, but rather extremely specific to what we did in the paper. Here is how we created a miscellaneous data table containing the genomic source of each sequence in an EcoPhylo analysis:
Show/Hide code to reproduce `genome-types.txt` file Oral microbiome dataset
DIR_PATH <- "REPRODUCIBLE_WORKFLOW_DATA"
# Im port default collection to get split names
DEFAULT_collection <- read_tsv(path(DIR_PATH, "collection-DEFAULT.txt"), col_names = c("representative", "collection")) %>%
mutate(representative = str_replace(pattern = "_split_00001",replacement = "", representative))
# Import cluster representatives and cluster members
detected_mmseqs_NR_cluster <- read_tsv(path(DIR_PATH, glue("{SCG}-mmseqs_NR_cluster.tsv")),
col_names = c("representative", "cluster_member")) %>%
filter(representative %in% DEFAULT_collection$representative)
# Import external-genomes.txt
genomes <- read_tsv(path(DIR_PATH, "external_genomes_MAGs_HOMD_Ribosomal_L19_detected.txt"))
# Make list of genomes categorized by their genome-type
REFG <- genomes %>% filter(grepl("GCA_", name))
MAG <- read_tsv("REPRODUCIBLE_WORKFLOW_DATA/ORAL_MAG_list.tsv", col_names = c("name"))
make_genome_type_file <- function(genomes, mmseqs_NR_cluster, SCG, REFG, MAG) {
genome_type_metadata <- mmseqs_NR_cluster %>%
separate(cluster_member, into = c("name", "number"), sep = glue("_{SCG}_"), remove = FALSE) %>%
dplyr::select(-number) %>%
mutate(genome_type = case_when(name %in% MAG$name ~ "MAG",
name %in% REFG$name ~ "REFG",
TRUE ~ "METAG")) %>%
dplyr::select(representative, genome_type) %>%
distinct() %>%
pivot_wider(id_cols = "representative", names_prefix = "genome_type.", names_from = "genome_type", values_from = "genome_type") %>%
mutate_at(vars(starts_with("genome_type")), ~ ifelse(!is.na(.x), TRUE, FALSE)) %>%
mutate(representative = str_c(representative, "_split_00001"))
genome_type_metadata_HQ_MAG <- mmseqs_NR_cluster %>%
separate(cluster_member, into = c("name", "number"), sep = glue("_{SCG}_"), remove = FALSE) %>%
dplyr::select(-number) %>%
mutate(HQ_MAG = case_when(name %in% MAG$name ~ TRUE,
TRUE ~ FALSE)) %>%
dplyr::select(representative, HQ_MAG) %>%
distinct() %>%
pivot_wider(id_cols = "representative", names_prefix = "HQ_MAG.", names_from = "HQ_MAG", values_from = "HQ_MAG") %>%
filter(HQ_MAG.TRUE == TRUE) %>%
select(-HQ_MAG.FALSE) %>%
mutate(representative = str_c(representative, "_split_00001"))
genome_type_metadata %>%
mutate("HQ_MAG.TRUE" = case_when(representative %in% genome_type_metadata_HQ_MAG$representative ~ TRUE,
TRUE ~ FALSE),
contains_genome = case_when(HQ_MAG.TRUE == TRUE | genome_type.REFG == TRUE ~ TRUE,
TRUE ~ FALSE),
metagenome_ONLY = case_when(genome_type.METAG == TRUE & genome_type.REFG == FALSE & HQ_MAG.TRUE == FALSE ~ TRUE,
TRUE ~ FALSE))
}
genome_type_metadata <- make_genome_type_file(genomes,
mmseqs_NR_cluster = detected_mmseqs_NR_cluster,
SCG = "Ribosomal_L19",
REFG = REFG,
MAG = MAG)
genome_type_metadata
Step 3. Import genome types misc data into anvi’o
anvi-import-misc-data genome_types.txt \
-p PROFILE.db \
--target-data-table items \
--just-do-it
Calculating genome recovery rate
To calculate genome recovery rates for any given taxon with EcoPhylo, we divided the number of sequence clusters that contained a sequence from a given genome recovery method by the total number of representative sequences EcoPhylo reported for that taxon. EcoPhylo taxonomically annotated ribosomal sequences with the program anvi-estimate-scg-taxonomy. You can extract this miscellaneous data along with a few other tables and calculate genome recovery rates like this:
anvi-estimate-scg-taxonomy
Step 1. Export items miscellaneous from EcoPhylo
This table will include all metadata from each representative ribosomal sequence and associated sequence cluster
anvi-export-misc-data -p PROFILE.db --target-data-table items -o items.tsv
Step 2. Calculate genome recovery rate from anvi-estimate-scg-taxonomy taxonomic annotations.
A caveat of anvi-estimate-scg-taxonomy is that it will not annotate ribosomal sequences that are x < 90% amino acid percent identity to it’s marker gene database. This will result in divergent ribosomal protein sequences not receiving a taxonomic annotation.
Here is an example of a function that will calculate the genome recovery rate if there is only one external genome type (e.g. MAGs) in your analysis.
find_MAG_recovery_rate <- function(item_additional_data, taxonomic_rank) {
name_taxa_rank <- taxonomic_rank
genome_recovery <- item_additional_data %>%
group_by(!!sym(taxonomic_rank)) %>%
summarize(total_detected = n(),
num_MAGs = sum(genomic_seq_in_cluster == "yes"),
MAG_recovery_rate = num_MAGs/total_detected) %>%
rename(taxa_name = !!sym(taxonomic_rank)) %>%
select(taxa_name, num_MAGs, total_detected, MAG_recovery_rate) %>%
arrange(desc(total_detected)) %>%
mutate(rank = name_taxa_rank)
return(genome_recovery)
}
taxonomic_ranks_GTDB <- c("t_domain",
"t_phylum",
"t_class",
"t_order",
"t_family",
"t_genus",
"t_species")
genome_recovery_rates_SCG_taxonomy_list <- map(taxonomic_ranks_GTDB, ~find_MAG_recovery_rate(item_additional_data, taxonomic_rank = .x))
Genome_recovery_rates_SCG_taxonomy <- bind_rows(genome_recovery_rates_SCG_taxonomy_list)
Taxonomic binning
To account for divergent ribosomal proteins that did not get a taxonomic assignment from anvi-estimate-scg-taxonomy, we used the ribosomal protein amino acid phylogenetic tree to group sequences with their surrounding clade members of the same taxa in the anvi-interactive interface and transferred their taxonomic annotation to the sequences with unknown classification - We call this taxonomic binning.
To recalculate genome recovery rates with taxonomic bins made from the EcoPhylo interactive interface the first step is to export the new collection of bins.
Step 1. Bin taxa of interest in the interactive interface and export the collections
Step 2. Export bins and items
anvi-export-misc-data -p PROFILE.db --target-data-table items -o items.tsv
anvi-export-collection -p PROFILE.db \
--collection-name bacteria_archaea \
--output-file-prefix bacteria_archaea_collection
Step 3. Calculate genome recovery rate with bins
DIR_PATH <- "REPRODUCIBLE_WORKFLOW_DATA"
# Example of bins for bacteria and archaea (great way to exclude the eukaryotic and plastid signal in the phylogenetic tree)
bacteria_archaea_collection <- read_tsv(path(DIR_PATH, "bacteria_archaea_collection.txt"), col_names = c("items", "bin"))
# Import the items misc data
item_additional_data_S15 <- read_tsv(path(DIR_PATH, "items.tsv"))
RP_S15_Bacteria_Archaea_recovery <- item_additional_data_S15 %>%
left_join(bacteria_archaea_collection) %>%
group_by(bin) %>%
summarize(total_detected = n(),
num_MAGs = sum(genomic_seq_in_cluster == "yes"),
MAG_recovery_rate = num_MAGs/total_detected)
Manually curate EcoPhylo tree
In our study, we constructed phylogenetic trees from large collections of ribosomal proteins, which in some cases included sequences from multiple domains of life. Additionally, many of these sequences originated from metagenomic assemblies, which are prone to artifacts. As a result, some of the phylogenetic trees displayed artificially long branches. Due to the high-throughput nature of the workflow and the potential for assembly artifacts, we manually inspected the trees, removed unusually long branches, and recalculated the phylogeny. Below are the basic steps to perform this process:
Step 1. Make collection of bad branches
In this step we manually examined the resulting phylogenetic trees for any branches that seemed suspicious then added them to a bin in the interactive interface.
Step 2. Export collection and remove those sequences from the ribosomal protein fasta file
HOME_DIR="ECOPHYLO" # Replace with EcoPhylo home directory
PROTEIN="" # Replace with name of protein from hmm_list.txt
cd $HOME_DIR
mkdir SUBSET_TREE
# Export default collection from EcoPhylo interactive interface
anvi-export-collection -p METAGENOMICS_WORKFLOW/06_MERGED/"${PROTEIN}"/PROFILE.db -C DEFAULT --output-file-prefix SUBSET_TREE/DEFAULT
# Export collection of denoting bad branches from EcoPhylo interactive interface
anvi-export-collection -C bad_branches -p METAGENOMICS_WORKFLOW/06_MERGED/"${PROTEIN}"/PROFILE.db --output-file-prefix SUBSET_TREE/bad_branches
# Clean fasta headers
cut -f 1 SUBSET_TREE/bad_branches | sed 's|_split_00001||' > SUBSET_TREE/bad_branches_headers.txt
anvi-script-reformat-fasta 02_NR_FASTAS/"${PROTEIN}"/"${PROTEIN}"-AA_subset.fa \
--exclude-ids bad_branches_headers.txt \
-o SUBSET_TREE/"${PROTEIN}"-AA_subset_remove_bad_branches.fa
ALIGNMENT_PREFIX=""${PROTEIN}"-AA_subset_remove_bad_branches"
# Calculate multiple sequence alignment
muscle -in SUBSET_TREE/"${PROTEIN}"-AA_subset_remove_bad_branches.fa \
-out SUBSET_TREE/"${ALIGNMENT_PREFIX}".faa -maxiters 1
# Trim alignment
trimal -in SUBSET_TREE/"${ALIGNMENT_PREFIX}".faa \
-out SUBSET_TREE/"${ALIGNMENT_PREFIX}"_trimmed.faa -gappyout
# Calculate tree
FastTree SUBSET_TREE/"${ALIGNMENT_PREFIX}"_trimmed_filtered.faa > SUBSET_TREE/"${ALIGNMENT_PREFIX}"_trimmed_filtered_FastTree.nwk
Step 3. Use anvi-split to remove bad branches from the EcoPhylo
interface
PROTEIN="" # Replace with name of protein from hmm_list.txt
grep -v -f SUBSET_TREE/bad_branches_headers.txt SUBSET_TREE/collection-DEFAULT.txt | sed 's|EVERYTHING|EVERYTHING_curated|' > SUBSET_TREE/my_bins.txt
anvi-import-collection SUBSET_TREE/my_bins.txt -C curated \
-p METAGENOMICS_WORKFLOW/06_MERGED/"${PROTEIN}"/PROFILE.db \
-c METAGENOMICS_WORKFLOW/03_CONTIGS/"${PROTEIN}"-contigs.db
anvi-split -C curated \
--bin-id EVERYTHING_curated \
-p METAGENOMICS_WORKFLOW/06_MERGED/"${PROTEIN}"/PROFILE.db \
-c METAGENOMICS_WORKFLOW/03_CONTIGS/"${PROTEIN}"-contigs.db \
--output-dir SUBSET_TREE/"${PROTEIN}"_curated
Step 4. Add the string “_split_00001” to each tree leaf so we can import it back into the interface
add_split_string_to_tree <- function(IN_PATH, OUT_PATH) {
# Import the phylogenetic tree from the specified input file
tree <- read.tree(IN_PATH)
# Create a dataframe containing the tree tip labels and append the split string
tree_tip_metadata <- tree$tip.label %>%
as_tibble() %>% # Convert tip labels to a tibble for easier manipulation
rename(tip_label = value) %>% # Rename the column to 'tip_label'
mutate(tip_label = str_c(tip_label, "_split_00001")) # Append '_split_00001' to each tip label
# Update the tree's tip labels with the modified labels
tree$tip.label <- tree_tip_metadata$tip_label
# Print the output path (optional; for debugging or logging purposes)
print(OUT_PATH)
# Write the modified tree to the specified output file in Newick format
write.tree(tree, file = OUT_PATH)
}
Step 5. Import new tree and visualize
TREE_NAME="FastTree_curated" # replace with description of tree
SCG="" # replace with protein name
anvi-import-items-order -p "${SCG}"_curated/EVERYTHING_curated/PROFILE.db \
-i "${SCG}"-AA_subset_remove_long_seqs_aligned_maxiters_2_trimmed_filtered_FastTree_ed.nwk \
--name $TREE_NAME
anvi-interactive -p "${SCG}"_curated/EVERYTHING_curated/PROFILE.db \
-c "${SCG}"_curated/EVERYTHING_curated/CONTIGS.db
EcoPhylo interactive interfaces
Here is how you can explore all of the EcoPhylo interactive interfaces created for this paper!
curl -L -o ECOPHYLO_INTERACTIVES_METHODS.tar.gz https://api.figshare.com/v2/file/download/53896007
tar -xvzf ECOPHYLO_INTERACTIVES_METHODS.tar.gz
cd ECOPHYLO_INTERACTIVES_METHODS
Here is the content of that directory:
$ tree
.
├── 00_README.md
├── HUMAN_GUT
│ ├── ECOPHYLO_WORKFLOW_human_gut_RP_L19
│ │ ├── AUXILIARY-DATA.db
│ │ ├── CONTIGS.db
│ │ └── PROFILE.db
│ ├── ECOPHYLO_WORKFLOW_human_gut_RP_S15
│ │ ├── AUXILIARY-DATA.db
│ │ ├── PROFILE.db
│ │ └── Ribosomal_S15-contigs.db
│ └── ECOPHYLO_WORKFLOW_human_gut_RP_S16
│ ├── AUXILIARY-DATA.db
│ ├── PROFILE.db
│ └── Ribosomal_S16-contigs.db
└── ORAL_CAVITY
├── ECOPHYLO_WORKFLOW_oral_cavity_RP_L19
│ ├── AUXILIARY-DATA.db
│ ├── PROFILE.db
│ └── Ribosomal_L19-contigs.db
├── ECOPHYLO_WORKFLOW_oral_cavity_RP_S15
│ ├── AUXILIARY-DATA.db
│ ├── PROFILE.db
│ └── Ribosomal_S15-contigs.db
└── ECOPHYLO_WORKFLOW_oral_cavity_RP_S2
├── AUXILIARY-DATA.db
├── PROFILE.db
└── Ribosomal_S2-contigs.db
Loading them up is really easy! Please note that the default state will automatically load the interfaces used in this paper.
Please note that the default EcoPhylo interative states below were overwritten after manually adjusting dimensions and colors. The majority of the time, the raw output of EcoPhylo will requiring manually adjustments by the user in anvi-interative to create an informative, beautiful representation of the underlying data.
rpL19 phylogeography in the human oral cavity
Here is an example for open the phylogeography of rpL19 across Shaiber et al. (2020). We also performed this analysis with rpS15, and rpS2. For all of the ORAL_CAVITY EcoPhylo interactive interface the default state will automatically load the interfaces used in this paper.
anvi-interactive -p ORAL_CAVITY/ECOPHYLO_WORKFLOW_oral_cavity_RP_L19/PROFILE.db \
-c ORAL_CAVITY/ECOPHYLO_WORKFLOW_oral_cavity_RP_L19/Ribosomal_L19-contigs.db

EcoPhylo analysis of rpL19 across the human oral cavity
rpS15 phylogeography in the human gut
Here is an example for open the phylogeography of rpS15 across Carter et al. (2023). We also performed the analysis with rpS16, and rpL19 which can be found in the figshare. For all of the ORAL_CAVITY EcoPhylo interactive interface the default state will automatically load the interfaces used in this paper.
anvi-interactive -p HUMAN_GUT/ECOPHYLO_WORKFLOW_human_gut_RP_S15/PROFILE.db \
-c HUMAN_GUT/ECOPHYLO_WORKFLOW_human_gut_RP_S15/CONTIGS.db

EcoPhylo analysis of rpS15 across human gut microbiome samples from the Hadza tribe
Contemporary genome collections massively under-represent microbial diversity in marine systems
In our study, we examined the phylogeography of ribosomal proteins across the global surface ocean ocean and a representative genome collection (MAGs, SAGs, and isolate genomes) to estimate genome recovery rates across this biome. We leveraged the anvi’o EcoPhylo workflow which is described in the methods paper above.
Reproducible / Reusable Data Products
The following data items are compatible with anvi’o version v8 or later. The anvi’o contigs-db and profile-db in them can be further analyzed using any program in the anvi’o ecosystem, or they can be used to report summary data in flat-text files to be imported into other analysis environments.
- DOI:10.6084/m9.figshare.28840841: Ribosomal protein interative profiling the open surface ocean (rpL14, rpS8, rpS11). To directly load this data, skip to the section EcoPhylo interactive interface of the open surface ocean
Set up environment
If you want to follow along and run each code block in the following reproducible, you will need to install anvi’o. Next you will need to download the associated data (DOI:10.6084/m9.figshare.28840898) and you are ready to go!
Load packages
These are the r packages you will need to load and install before running the analysis:
packages <- c("tidyverse", "ggpubr", "fs", "ape", "treeio", "glue", "plotly", "readxl")
suppressWarnings(suppressMessages(lapply(packages, library, character.only = TRUE)))
The majority of commands using anvi-run-workflow were run on high performance compute clusters (HPC) leveraging the powerful SLURM wrapper clusterize. Without access to compute nodes, these commands would take a VERY long time. Please check out this blogpost for more information: Running workflows on a cluster. All BASH commands in this reproducible workflow will need to be modified to run on your own HPC system. See below for an example some modifiable paramaters that might be helpful.
Selecting ribosomal proteins
Before running the EcoPhylo workflow, we selected a subset of ribosomal proteins—rpL14, rpS8, and rpS11—to profile the surface ocean. These proteins were chosen because they appeared in single-copy across the majority of genomes in our dataset. Sadly, genome types such as metagenomc-assembled genomes and single amplified genomes are rarely completely reconstructed thus, we potentially cannot profile an entire genome collection at the same time. We next ensured these ribosomal proteins were not systematically over- or under-assembled relative to other ribosomal proteins across the surface ocean metagenomic assemblies.
For more examples in our methods paper you can navigate here: Identifying the most frequent ribosomal proteins in a genomic collection
To visualize these distributions, here is a code snippet:
Step 1. Run EcoPhylo workflow to annotate surface ocean genome dataset with Bacteria_71 SCG collection
Here is an “example” of prototypical command with some basic descriptions of the various parameters. Make sure to fill in everything on your own :)
anvi-run-workflow -w ecophylo \ # anvio worklow name
-c config.json \ # workflow config file
--additional-params \ # extract parameters that go straight to snakemake
--cluster "qsub {threads}" \ # example string to send jobs to HPC
--jobs=120 \ # number of co-occuring jobs
--resources nodes=120 \ # num. of nodes the workflow can use
--latency-wait 100 \ # wait 100 sec. for output file to appear
--keep-going \ # finish as many jobs as possible even if one job fails
--rerun-incomplete # restart any jobs that failed
Step 2. Find SCGs that detect the majority of the genomic dataset
Here we used anvi-script-gen-hmm-hits-matrix-across-genomes to extract the matrix of hmm-hits from both the metagenomic assemblies and genome collection.
# Extract SCGs across metagenome dataset
anvi-script-gen-hmm-hits-matrix-across-genomes -e metagenomes.txt \
--hmm-source Bacteria_71 \
-o Bacteria_71_METAGENOME_MATRIX.txt
# Extract SCGs genome collection dataset
anvi-script-gen-hmm-hits-matrix-across-genomes -e external_genomes.txt \
--hmm-source Bacteria_71 \
-o Bacteria_71_GENOME_MATRIX.txt
Step 3. Plot ribosomal protein detection across metagenomic assemblies
Here we imported the matrices produced in the previous step to visualize the copy number distribution:
DIR_PATH <- ""
Bacteria_71_GENOME_MATRIX <- read_tsv(path(DIR_PATH, "TABLES/Table_SI01.tsv"))
# set up a pretty theme for the plots
SCG_detection_plot_theme <- theme_light() +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1, family = "Helvetica Neue Condensed Bold"),
axis.ticks.x = element_blank(),
legend.title = element_blank(),
axis.text = element_text(size = 6, family = "Helvetica Neue Condensed Bold"),
axis.title = element_text(size = 6, family = "Helvetica Neue Condensed Bold"),
text = element_text(size = 6, family = "Helvetica Neue Condensed Bold"),
plot.title = element_text(hjust = 0.5, size = 8, family = "Helvetica Neue Condensed Bold"))
plot_metagenome_SCGs <- function(X, YLAB, XLAB, TITLE) {
X_long <- X %>%
pivot_longer(-genome_or_bin) %>%
rename(SCG = "name") %>%
group_by(SCG) %>%
summarize(total = sum(value))
mean_SCG_count <- X_long$total %>% mean() %>% round()
X_long %>%
ggplot(aes(x = total, y = fct_reorder(SCG, total))) +
geom_point() +
scale_x_continuous(labels=scales::comma) +
geom_vline(xintercept = mean_SCG_count, color = "blue") +
SCG_detection_plot_theme +
ylab(YLAB) +
xlab(XLAB) +
ggtitle(glue("Single-copy core gene frequencies across {TITLE} (n = {X$genome_or_bin %>% length()}; mean = {mean_SCG_count})"))
}
plot_metagenome_SCGs_frequencies <- plot_metagenome_SCGs(Bacteria_71_METAGENOME_MATRIX,
YLAB = "Single-copy core genes (anvi'o Bacteria_71)",
XLAB = "Total SCG counts",
TITLE = "surface ocean metagenomes")
Step 4. Plot ribosomal protein detection across genome dataset
DIR_PATH <- ""
Bacteria_71_GENOME_MATRIX <- read_tsv(path(DIR_PATH, "TABLES/Table_SI01.tsv"))
plot_genome_SCGs <- function(X, TITLE, XLAB, YLAB) {
SCG_MATRIX_long <- X %>% pivot_longer(-genome_or_bin) %>% rename(SCG = "name")
# Get order of bar charts
percent_order <- X %>%
pivot_longer(-genome_or_bin) %>%
rename(SCG = "name") %>%
mutate(presence = case_when(value >= 1 ~ 1,
value < 1 ~ 0)) %>%
group_by(SCG) %>%
summarize(total = sum(presence)) %>%
mutate(percent = (total/SCG_MATRIX_long$genome_or_bin %>% unique() %>% length())*100)
num_scgs <- SCG_MATRIX_long %>% .$SCG %>% unique() %>% length()
num_genomes <- SCG_MATRIX_long %>% .$genome_or_bin %>% unique() %>% length()
SCG_MATRIX_long %>%
group_by(SCG) %>%
count(value) %>%
mutate(percent_occ = n/sum(n)*100) %>%
mutate(value = as.character(value),
once = n[value == "1"]) %>%
left_join(percent_order) %>%
ggplot(aes(x = fct_reorder(SCG, percent), y = percent_occ, fill = value)) +
geom_bar(stat="identity") +
SCG_detection_plot_theme +
theme(legend.position="left") +
ylab(XLAB) +
xlab(YLAB) +
ggtitle(glue("Single-copy core gene frequencies across {TITLE} (n = {X$genome_or_bin %>% length()})"))
}
plot_bacteria_SCG_frequency <- plot_genome_SCGs(Bacteria_71_GENOME_MATRIX,
YLAB = "Single-copy core genes (anvi'o Bacteria_71)",
XLAB = "Percent of genome collection detected",
TITLE = "surface ocean genomes")
# Plot both plots at the same time!
ggarrange(plot_bacteria_SCG_frequency, plot_metagenome_SCGs_frequencies, ncol = 2)

Single-copy core genes detected across surface ocean genomic and metagenomic assembly datasets.
EcoPhylo interactive interface of the open surface ocean
Post-processing EcoPhylo analysis
Prior to investigating the results of phylogeography of ribosomal proteins across the surface ocean, we took multiple post-processing steps. Here are links to the methods paper described above which go into these steps in detail:
- Removing genomes that are not detected in metagenomic samples
- Calculating genome recovery rate
- Manually curate EcoPhylo tree: In this study, we calculated phylogenetic trees of large collections with thousands of ribosomal proteins which included sequences from all domains of life, including plastids and mitochondria, which in some cases yielded unusually long branches. This required manual curation, pruning, and tree recalculation to remove metagenome assembly artifacts and plastid/mitochondria ribosomal proteins, we manually inspected and removed and recalculated the tree. You can read more details about this methodology here: Manually curate EcoPhylo tree.
Visualize ribosomal protein phylogeography with EcoPhylo
Here is how you can explore all of the EcoPhylo interactive interfaces created for this paper!
curl -L -o ECOPHYLO_INTERACTIVES_SURFACE_OCEAN.tar.gz https://api.figshare.com/v2/file/download/53897114
tar -xvzf ECOPHYLO_INTERACTIVES_SURFACE_OCEAN.tar.gz
cd ECOPHYLO_INTERACTIVES_SURFACE_OCEAN
Here are the contents of that directory:
$ tree
.
├── 00_README.md
├── SURFACE_OCEAN
├── ECOPHYLO_WORKFLOW_surface_ocean_RP_L14
│ ├── AUXILIARY-DATA.db
│ ├── CONTIGS.db
│ └── PROFILE.db
├── ECOPHYLO_WORKFLOW_surface_ocean_RP_S11
│ ├── AUXILIARY-DATA.db
│ ├── CONTIGS.db
│ └── PROFILE.db
└── ECOPHYLO_WORKFLOW_surface_ocean_RP_S8
├── AUXILIARY-DATA.db
├── CONTIGS.db
└── PROFILE.db
Please note that the default EcoPhylo interative states below were overwritten after manually adjusting dimensions and colors. The majority of the time, the raw output of EcoPhylo will requiring manually adjustments by the user in anvi-interative to create an informative, beautiful representation of the underlying data.
Here is how to open main figure of the manuscript which represents the phylogeography of rpL14 across the surface ocean. We also performed the analysis with rpS8, and rpS11 which can be found in the figshare directory. For all EcoPhylo interfaces, the default state will show all of the metagenomes used in the paper. If you would like to filter for only for only metagenomes with x > 50 million reads to replicate the main figures in the text, please load the deep_sequencing state.
anvi-interactive -p SURFACE_OCEAN/ECOPHYLO_WORKFLOW_surface_ocean_RP_L14/PROFILE.db \
-c SURFACE_OCEAN/ECOPHYLO_WORKFLOW_surface_ocean_RP_L14/CONTIGS.db

EcoPhylo analysis of rpL14 across the global surface ocean
If you load the deep_sequencing state, you will reproduce the figure in the main text before polishing, which will allow you to interactivel explore the dataset:
anvi-interactive -p SURFACE_OCEAN/ECOPHYLO_WORKFLOW_surface_ocean_RP_L14/PROFILE.db \
-c SURFACE_OCEAN/ECOPHYLO_WORKFLOW_surface_ocean_RP_L14/CONTIGS.db \
--state-autoload deep_sequencing

EcoPhylo analysis of rpL14 across the global surface ocean subsetted for metagenomes with x > 50 million reads
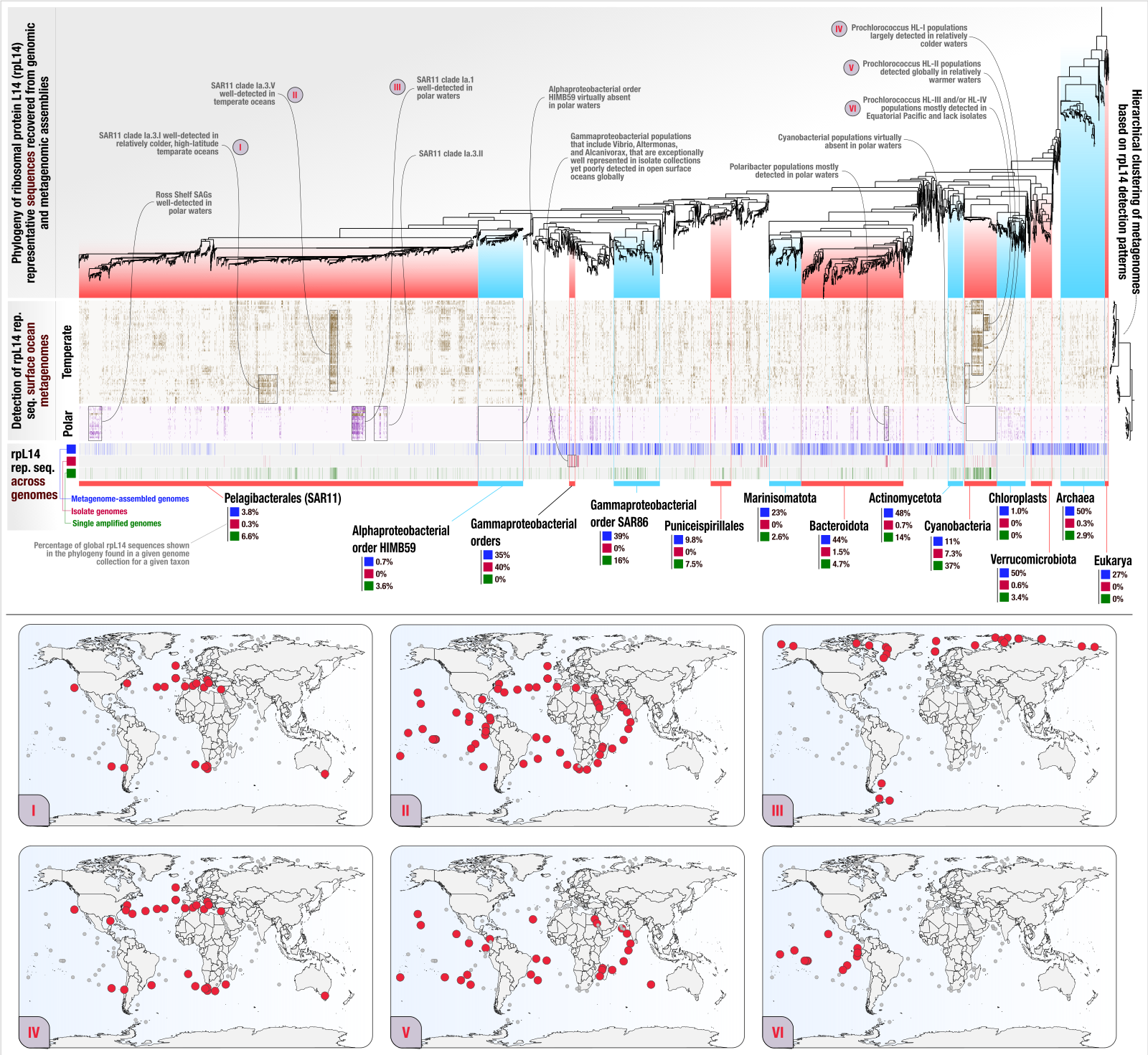
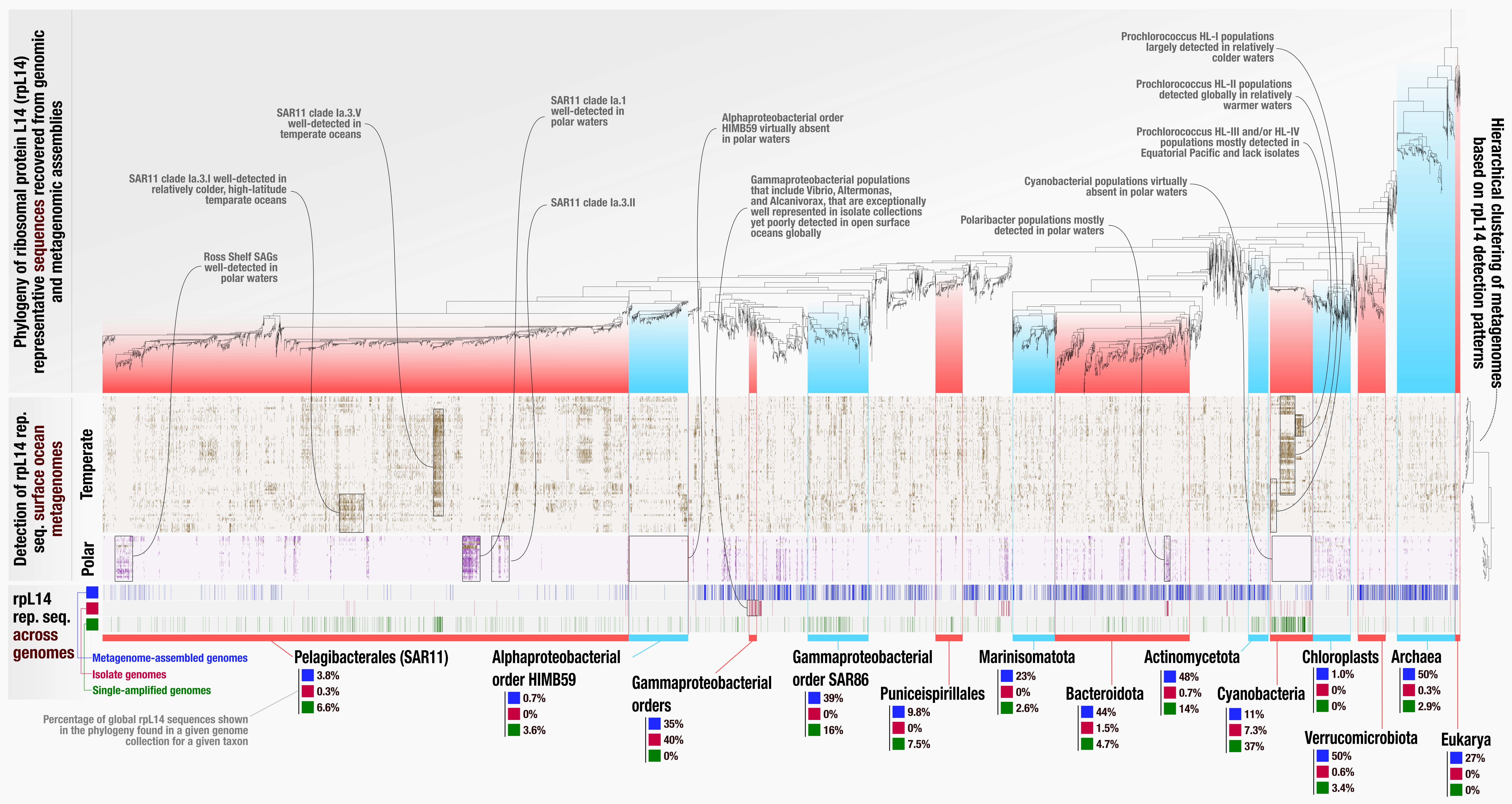
After acquiring the SVG version of the display shown above from anvi’o, we used Inkscape to polish it for publication, which gave us the following image:

EcoPhylo analysis of rpL14 across the global surface ocean, polished. If you would like to see this image in its full resolution, you can right-click this link and open it in a new window.
{kind=link}
Scatter plot of the number of SCGs recovered in assembled metagenomes vs sequencing depth from 237 surface ocean metagenomes.
Metagenome sequencing depth can potentially impact the number of populations that can be assembled in genome-resolve metagenomics. To explore this, we looked at the relationship between sample read depths and the number of single-copy core genes detected in associated sample metagenomic assemblies.
# load data
DIR_PATH <- ""
reads_SCGs <- read_tsv(path(DIR_PATH, "TABLES/Table_SI04.tsv"))
# Fit a linear model
model <- lm(num_SCG ~ total_num_reads, data = reads_SCGs)
summary(model)
# Extract p-value and R-squared
p_value <- summary(model)$coefficients[2, 4] # p-value for the latitude term
r_squared <- summary(model)$r.squared
slope <- round(coef(model)[2], 5) # Slope for abs_lat
# Assign sample colors
surface_ocean_project_colors <- c(TARA = "#E79B1B", TARA_arctic = "#9b00dd", BGEO = "#00c438", HOTS ="#ff0600", BATS = "#ff0600", Malaspina = "yellow", Pachiadaki = "Dark blue", Ross_Shelf = "Black") # Sample colors
# plot lm
num_SCG_sequencing_depth_lm_p <- reads_SCGs %>%
mutate(reads_millions = total_num_reads / 1e6) %>%
ggplot(aes(x = reads_millions, y = num_SCG, , group = interaction(Project, size_fraction))) +
geom_point(aes(shape = size_fraction, fill = Project), size = 4, color = "black") +
scale_shape_manual(values = c(21, 22, 24)) +
scale_fill_manual(name = "Project", values = surface_ocean_project_colors[!names(surface_ocean_project_colors) %in% c("Pachiadaki", "Ross_Shelf")]) +
geom_smooth(aes(group = 1), method = "lm", se = TRUE) +
annotate("text", x = 50, y = 20000, label = paste("p-value:", round(p_value, 3), "\nR²:", round(r_squared, 3), "\nSlope:", slope),
hjust = 1, size = 5) +
xlab("Metagenomic sequencing depth (millions of reads)") +
ylab("Number of single-copy genes detected (Bacteria_71 marker set)") +
ggtitle("Recovery of Single-Copy Genes Scales with Metagenomic Sequencing Depth") +
theme_minimal() +
theme(plot.title = element_text(hjust = 0.5),
legend.position="bottom") +
scale_y_continuous(labels = scales::comma)
num_SCG_sequencing_depth_lm_p

Scatter plot of the number of SCGs recovered in assembled metagenomes vs sequencing depth from 237 surface ocean metagenomes.
Regression analysis of latitude versus bacterial + archaeal rpL14 richness from surface ocean metagenomes
Here we examined the relationship between microbial community richness and latitude in the global surface ocean using ribosomal protein phylogeography.
DIR_PATH <- ""
data_deep_metagenomes <- read_tsv(path(DIR_PATH, "TABLES/Supplementary_Table_03.tsv"))
model <- lm(richness ~ abs_lat, data = data_deep_metagenomes)
summary(model)
# Extract p-value and R-squared
p_value <- summary(model)$coefficients[2, 4] # p-value for the latitude term
r_squared <- summary(model)$r.squared
slope <- round(coef(model)[2], 2) # Slope for abs_lat
lat_richness_lm_p <- data_deep_metagenomes %>%
ggplot(aes(x = abs_lat, y = richness, fill = Project)) +
geom_point(shape = 21, size = 2) +
geom_smooth(aes(group = 1), method = "lm", se = TRUE) +
annotate("text", x = 75, y = 800, label = paste("p-value:", round(p_value, 15), "\nR²:", round(r_squared, 3), "\nSlope:", slope),
hjust = 1, size = 5) +
labs(x = "Absolute Latitude", y = "rpL14 Richness", title = "rpL14 richness vs. Absolute Latitude") +
theme_minimal() +
theme(legend.position="bottom")
lat_richness_lm_p

Regression analysis of latitude versus bacterial + archaeal rpL14 richness from surface ocean metagenomes
Log mean Q2Q3 coverage of bacteria and archaea rpL14 sequences by genome type across surface ocean.
Relative abundance of a microbe can potentially impact it’s ability to be recovered as a genome. We explore this relationship across metagenome-assembled genomes, single amplified genomes, and isolate genomes using rpL14 phylogeography.
DIR_PATH <- ""
data <- read_tsv(path(DIR_PATH, "TABLES/Supplementary_Table_06.tsv"))
data$log_mean_cov <- log10(data$mean_cov) # convert data to log10
aov_model <- aov(log_mean_cov ~ genome_type, data = data)
summary(aov_model)
TukeyHSD(aov_model)
box_plot_stats <- function(data, column, label.x, label.y, title, y_title){
# Run ANOVA and Tukey HSD
formula <- as.formula(paste(column, "~ genome_type"))
aov_result <- aov(formula, data = data)
tukey_result <- TukeyHSD(aov_result)
# Extract p-values from Tukey test
tukey_pvals <- tukey_result$genome_type[, "p adj"]
# Create the basic plot
p <- ggboxplot(data, x = "genome_type", y = column,
# yscale = "log10",
add = "jitter",
# fill = "genome_type",
# palette = "jco"
) +
# Global ANOVA p-value
stat_compare_means(method = "anova", label.x = label.x, label.y = label.y) +
theme(plot.title = element_text(hjust = 0.5),
legend.position="none") +
labs(title = title,
x = "Genome Recovery Method",
y = y_title)
# Add annotations for Tukey HSD results
y_pos <- max(data[[column]], na.rm = TRUE) * c(1.2, 1.5, 1.8) # Adjust these values
# Add brackets and p-values manually
p <- p +
geom_bracket(xmin = "MAG", xmax = "SAG", y.position = y_pos[1],
label = paste("p =", round(tukey_pvals["SAG-MAG"], 15))) +
geom_bracket(xmin = "MAG", xmax = "NONE", y.position = y_pos[2],
label = paste("p =", round(tukey_pvals["NONE-MAG"], 15))) +
geom_bracket(xmin = "SAG", xmax = "NONE", y.position = y_pos[3],
label = paste("p =", round(tukey_pvals["SAG-NONE"], 15)))
return(p)
}
box_plot_stats(data = data,
column = "log_mean_cov",
label.x = 3.8,
label.y = 3,
title = "Log-MEAN Coverage by Genome Type (ANOVA and TukeyHSD)",
y_title = "Mean Coverage (log10 scale)")

Log mean Q2Q3 coverage of bacteria and archaea rpL14 sequences by genome type across surface ocean.