



Table of Contents
The methods paper for oligotyping appeared in Methods in Ecology and Evolution:

It was somewhat cumbersome to decide which journal would be the most appropriate to submit this paper. We hoped to publish it in a journal that is focused more on the ecology than bioinformatics. Dr. Arpat Ozgul suggested us to consider MEE, which is a journal that “promotes the development of new methods in ecology & evolution, and facilitates their dissemination and uptake by the research community“. We took his advice, and I am very glad that they accepted to review a paper that describes a method mostly for microbial ecologists. I think MEE deserves more attention from scientists who study microbial ecology.
In this paper we demonstrated the efficacy of oligotyping using three datasets, and I believe there is finally a clear and formal explanation of what oligotyping offers.
Here is a lovely story of two most abundant Pelagibacter oligotypes we recovered from Little Sippewissett Marsh, representative sequences of which are 99.57% similar to each other (Fig. 3a in the paper):
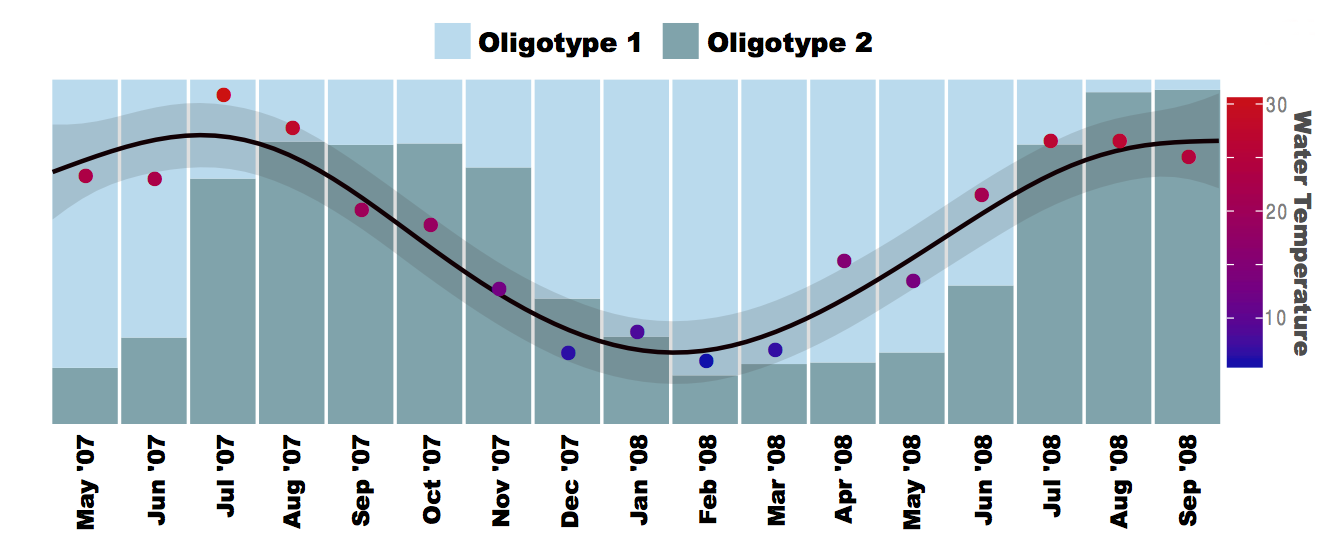
Their relative abundance change with the changing temperature over the course of 17 months; a recovery which would have been impossible to be made with taxonomy or OTU clustering at any threshold below 99.6%.
There is more:
- Oligotype 1 is identical to the 16S rRNA of Candidatus Pelagibacter ubique HTCC1062 (strain 1) over the sequenced region, and Oligotype 2 is identical to the 16S rRNA of Pelagibacter strain HTCC7211 (strain 2). These are two Pelagibacter organisms whose genomes are fully sequenced despite all the difficulties with cultivation thanks to the efforts of Giovannoni and his colleagues.
- Thanks to the 2012 study done by Mark V. Brown and his colleagues, we know that while strain 1 predominates polar regions, while strain 2 predominates tropical regions. In line with that observation, we see oligotype 1 more in colder months.
So these are very similar organisms at 16S rRNA gene level, however they are adapted to different temperature zones, and this information is right in there if we pay attention to very subtle nucleotide variations.
This is yet another example that reminds us what is essentially wrong in the general approach of measuring phylogenetic distances using 16S. “Well, it works most of the time” you might say, referring all those beautiful PCA plots showing us how different soil, feces and ocean samples are from each other, and I would concur! But microbial ecologists are rarely interested in comparing feces to ocean, and although a lot of success stories this field recovered through 16S-based surveys, I think it may not be a very bad idea to go back and look into studies where we didn’t see much of a difference between our control and treatment groups with respect to taxa, OTUs, or ecotypes.