
Table of Contents
I am very pleased to announce that Frontiers in Microbiology is now hosting a research topic on oligotyping, which is open for submissions!
This is what I had said on these pages about a year ago. Today, the research topic New insights into microbial ecology through subtle nucleotide variation is almost concluded, and contains 8 publications. The common theme among all these publications is that they use oligotyping.
I thought this would be a good time to offer a glimpse of what has been published in this collection so far.
Gut microbiomes of cheetahs and jackals

In their study Menke et al. compare the gut microbiomes of two sympatric mammalian carnivores, cheetah and black-backed jackal, by sequencing the V4 region of the 16S rRNA gene. They amplified the material for sequencing from fecal samples of free-ranging animals, too!
Being sympatric, and having somewhat similar diets, these animals seem to have similar gut microbiomes in the big picture, yet their microbiomes are different enough at lower levels of taxonomy for them to separate from each other on an ordination. The authors show that even genera that seem to be shared among the two species are in fact composed of different oligotypes. For instance Blautia is pretty abundant microbial genus in both cheetah and jackal group, yet Blautia oligotypes differ dramatically between the two (cheetah samples are on the left) (Figure 6 from Menke et al.):
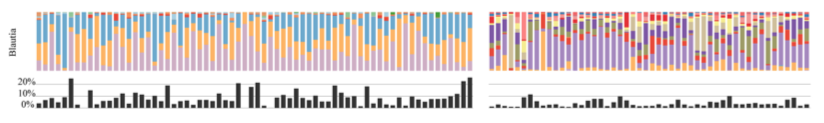
The figure shows that Blautia group is more abundant in cheetah samples in general, which is how it contributes to the separation of cheetah samples from jackal samples on an ordination. But with oligotypes, beyond separation, it is possible to recover specific microbial markers to distinguish each group from the other as it seems some Blautia organisms exclusively occur in sampls coming from one species or the other. I am especially interested in Blautia because this genus seems to be a great marker for different host species, as we had shown in our 2014 ISMEJ publication, and previously shown by Sandra et al. in an Environmental Microbiology paper, again, using oligotyping.
But Blautia is not the only genus with oligotypes that distribute differently between the two species. Here is another example, Slackia:
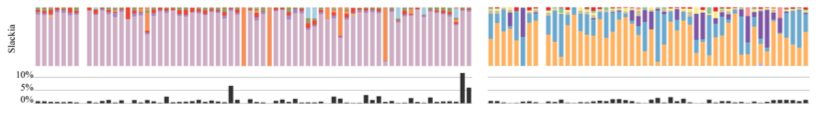
You can read more of this study here: doi:10.3389/fmicb.2014.00526
Arcobacter in sewage

To better understand the ecological factors that affect the survival and growth of Arcobacter spp. in sewer infrastructure, Fisher et al. dissect the Arcobacter group (which comprise 5% to 11% of sewage bacterial community) into a more precisely-defined taxonomic units by oligotyping reads coming from V4V5 region of the gene.
The study contains sequencing data from 12 sewage treatment centers in the US that were sampled three times: August 2012, January 2013, and April 2013 (there is also one additional sample from Spain). The stability of sewage systems is just very impressive. The first figure in the study shows that although the composition of Arcobacter oligotypes can differ from one station to the other, they do not differ as much across the three time points within one station (even when their abuncance in the the overall sample they are found change quite drastically):
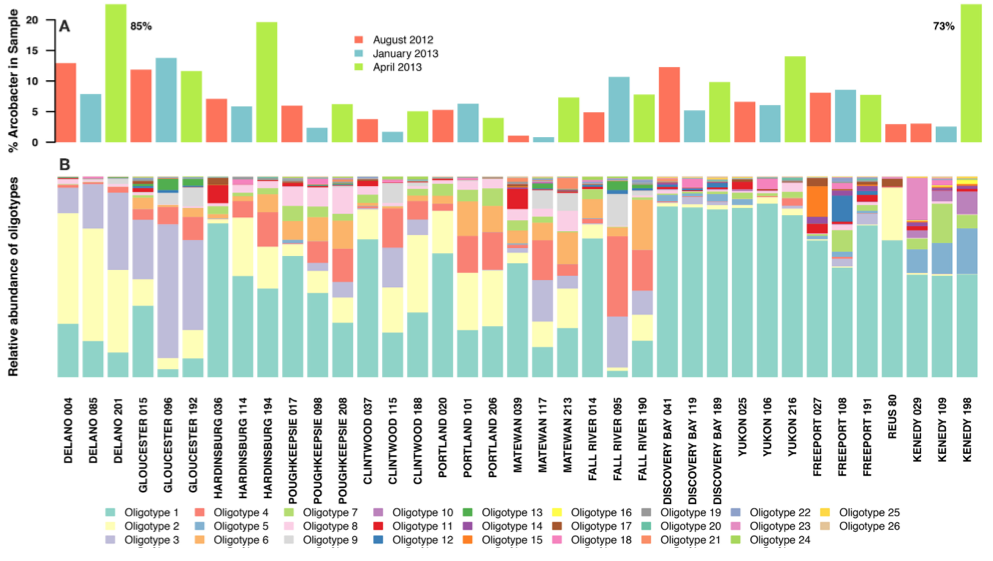
The study has an extensive discussion on factors that affect the growth of Arcobacter in sewage, and the temperature, as usual, is one key player:
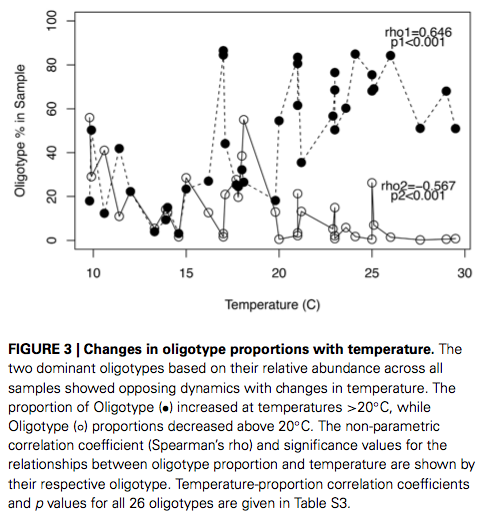
You can read more of this study here: doi: 10.3389/fmicb.2014.00525
Dynamics of tongue microbial communities

In this study, which I was a part of, Mark Welch et al. re-analyzes the infamous microbiome time series data published by Caporaso et al.. We had recently re-analyzed and published the oral samples collected by the HMP project. The HMP data were mostly representing a cross-sectional sampling of a large group of individuals. Cross-sectional sampling is great as they cover a great number of different individuals, but the lack of multiple samples from each individual leaves many other questions ‘unanswerable’. For instance one of the interesting findings we had in our previous study was this (from Fig 4 in Eren et al.):
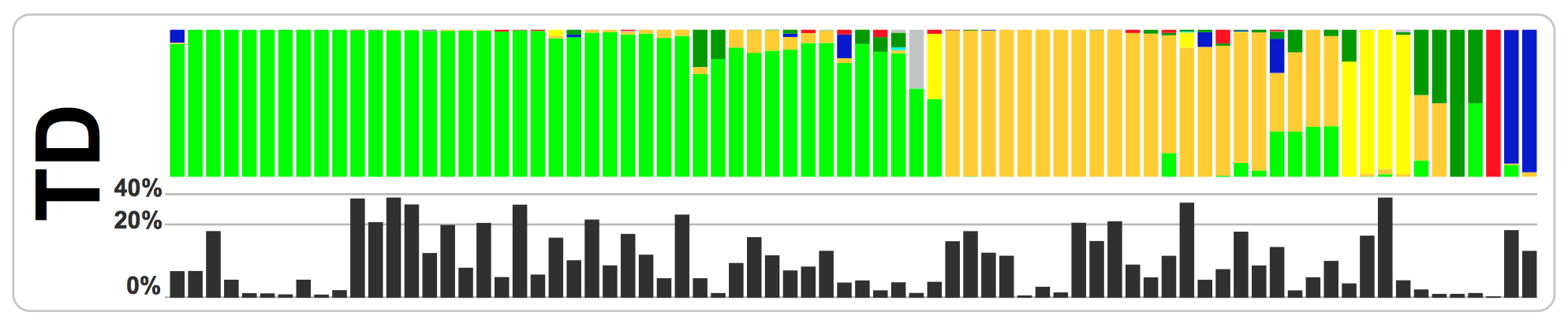
In this figure each bar represents one individual, and colors show the distribution of different Neisseria oligotypes in a person’s tongue. What is interesting about this figure is that each individual is dominated by one of the Neisseria oligotypes (almost all of which are 99% identical to each other at the sequenced region and would have been binned together by conventional methods). So, what is going on here? Are these oligotypes represent functionally identical organisms? Do clusters of colors identify different stable community states in the oral cavity? Are some of those samples that seem to have multiple colors examples of transient community states? If we had looked at one of those individuals for a long period of time, would have we seen a sudden switch in the Neisseria population from one state to another? And most importantly, what governs these patterns? Neutral effects? The host immune system? Diet? Smoking or drinking habits? The rest of the microbiome? All of the above? Or none?
The dataset published in Caporaso et al.’s study, which contains 396 time points from only two individuals of course was a natural follow-up to our previous analysis of the HMP in an attempt to answer some of these questions. So. Jessica Mark Welch and Daniel Utter did the analysis, and here is the distribution of Neisseria oligotypes identified in these two individuals:
I wish colors were identical to the previous study, but sequencing different regions of the 16S rRNA gene makes it very very hard to connect organisms to each other confidently.
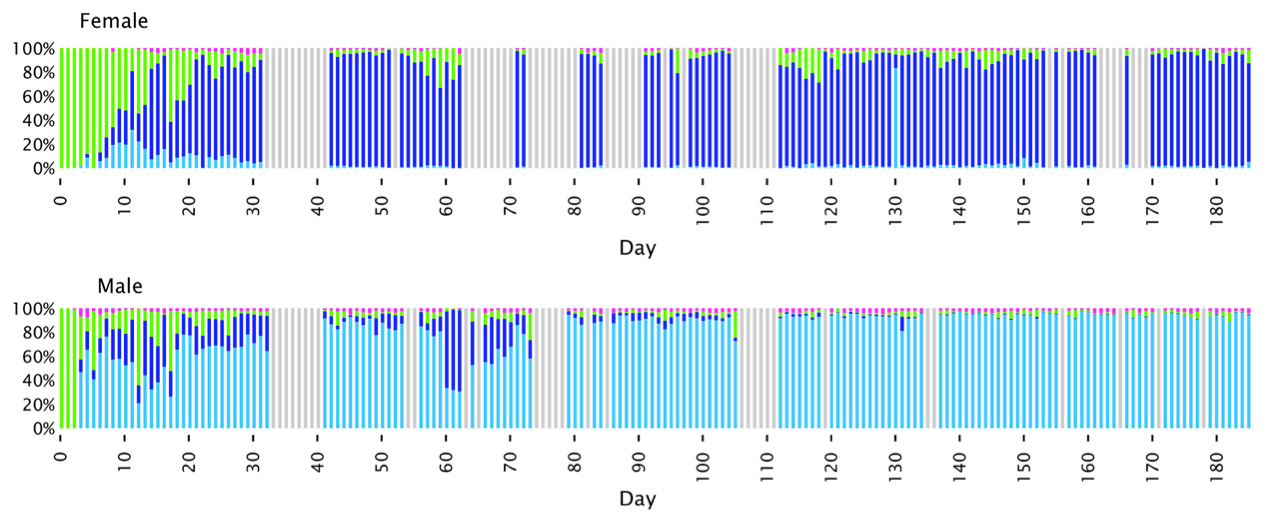
There are a number of interesting things going on in this figure. For instance, at the very beginning of the sampling period, each individual is composed of one the Neisseria oligotype shown in green. Then they diverge into their own type, blue for female and cyan for male, and they stay like that! As a note, blue and cyan are one nucleotide apart from each other, so this pattern would also have been lost if conventional OTU clustering had been used to analyze the dataset. Here is a quote from the paper talking about this figure:
These dynamics display two main characteristics which, taken together, may be termed a phase transition. The major behavior is one of stability. For most of the time, the oligotype distribution within an individual was essentially invariant, irrespective of whether the dominant oligotype in the individual was [Cyan] or [Blue]. The second property was of abrupt transition to an alternate oligotype. The time series data showed several instances in which a community initially dominated by one oligotype became transiently mixed and then transitioned to a state where one oligotype was dominant. These properties suggest that the evenly mixed populations of Neisseria on the tongue found in some individuals in the HMP data [shown in the previous figure] are transient states. Occasional replacement of the dominant oligotype argues against strong founder effects and priority effects for this taxon in the tongue microbiota. Throughout these transitions the fourth oligotype, [Purple], did not participate in the apparently competitive or exclusionary dynamics of types [Cyan] and [Blue], but persisted in relatively stable proportion in the community, likely demonstrating a subdivision of functional/ecological roles even among these very closely related taxa.
You can read the rest of the study here: doi: 10.3389/fmicb.2014.00568
An R package for the entropy decomposition

One of the great surprises of this collection was to see Alban Ramette and Pier Luigi Buttigieg’s R implementation of oligotyping and Minimum Entropy Decomposition. The GitHub repository for ‘otu2ot’ is located here: https://github.com/aramette/otu2ot.
Ramette et al.’s R library makes it much easier for R users to start using the approach on their datasets. The R library not only almost completely matches the functionality of a full oligotyping installation, but it also comes with two novel features: Broken stick model, to identify which oligotypes are more abundant than expected by chance, and a one-pass procedure, to rapidly assess the amount of microdiversity present in a group of sequences after only one round of entropy calculation.
The oligotyping pipeline comes all sorts of bells and whistles in an attempt to improve the user experience. The R implementation, on the other hand, is more likely to be used by statisticians and developers to test and improve the approach. Broken stick model is a great example to that, and opens up a great path to better and statistically sound noise filtering on this type of data.
You can read the study here: doi: 10.3389/fmicb.2014.00601
And others
There are four other publications in the collection, one of which is still in press as of today:
-
Oligotyping reveals community level habitat selection within the genus Vibrio
Victor T. Schmidt, Julie Reveillaud, Erik Zettler, Tracy J. Mincer, Leslie Murphy and Linda A. Amaral-Zettler. -
”Phaeocystis antarctica blooms strongly influence bacterial community structures in the Amundsen Sea polynya“
Tom O. Delmont, Katherine M. Hammar, Hugh W. Ducklow, Patricia L. Yager and Anton F. Post. -
”Biogeographic patterns of bacterial microdiversity in Arctic deep-sea sediments (HAUSGARTEN, Fram Strait)“
Pier Luigi Buttigieg and Alban Ramette. -
”Oligotyping reveals stronger relationship of organic soil bacterial community structure with N-amendments and soil chemistry in comparison to that of mineral soil at Harvard Forest, MA, USA“
Swathi Anuradha Turlapati, Rakesh Minocha, Stephanie Long, Jordan Ramsdell and Subhash C Minocha.
Final Words
I am very thankful for everyone who contributed to this collection.
The diversity of environments studied in these publications, and the rate of the recovery of ecologically meaningful findings show that there is a great potential for highly resolved depictions of microbiomes.
Oligotyping and minimum entropy decomposition provided a framework for researchers to explore these dimensions of their datasets at levels of single-nucleotide resolution. I am sure there will be many similar approaches going forward, and a lot of research will take place to make these results more accurate, and to utilize them in searching for answers to outstanding questions in microbial ecology.
It is just about getting the mindset of microbial ecology out of the local minimum it stuck called “3%”. The rest will come very quickly.
This has been a great experience on many levels, and I am very thankful to Frontiers in Systems Microbiology and the great team behind it for this opportunity.
I think research topics in Frontiers provide a great framework to create coherent collections of work that aim to contribute to a particular, well-defined issue.
If you think you have a study that would fit well into this collection, please send me or microbiology.researchtopics at frontiersin dot org an e-mail.