
Table of Contents
- Downloading and characterizing Kowarsky et al. contigs
- Sequences of ribosomal proteins
- Searching ribosomal proteins in NCBI’s nr database
- Some speculations
- Hypothesis: Oral cavity may be the actual reservoir for this CPR (Concluision: Not likely)
- Clifford Beall reports back: no hits from 20 saliva metagenomes
- Meren confirms: no hits from 14 plaque and tongue metagenomes
- Functions
- Can we say it though?
Twitter is bad. I mostly follow scientists, and often end up running into interesting findings from other groups that make me want to take a quick look at their data. Although most of our procrastinations don’t end up on the blog, sometimes they do: 1, 2, 3. Well, today was one of those days.

The study above reveals microbial findings from the human blood with a potential to start some debates about contamination.
Briefly, Kowarsky et al. take hundreds of blood samples collected from tens of patients, and use shotgun seqeuncing and assembly strategies to recover contigs from cell-free DNA. They remove sequences that match to the human genome, and investigate what is there in the remaining contigs. The authors also validate some of their findings by performing independent bench experiments, which is very nice to see since unfortunately ‘omics findings are rarely validated by additional experiments.
While you may be asking yourself whether some of the findings may have been driven by contamination, I am looking at the authors’ use of the term “dark matter” in their manucript and ask myself when should I move to the countryside and raise chickens. But that’s just me.
Interesting findings in this study that are summarized in its title intrigued me to take another look at their assembly. I was very glad to learn that the authors did make available not only the short reads for metagenomes, but also their assembly results. I thank them very much for their observance of open science practices (which is something that is also not as common as it should be).
Downloading and characterizing Kowarsky et al. contigs
I started by downloading the assembled contigs, and created an anvi’o contigs database. Everything took about 2-3 minutes:
# download the assembled contigs
curl -L http://www.pnas.org/content/suppl/2017/08/21/1707009114.DCSupplemental/pnas.1707009114.sd05.txt -o Kowarsky_et_al.fa
# generate an anvi'o contigs database
anvi-gen-contigs-database -f Kowarsky_et_al.fa \
-o Kowarsky_et_al.db \
--name 'Kowarsky et al contigs'
# run anvi'o's default HMM profiles on this contigs database
# using 10 cores
anvi-run-hmms -c Kowarsky_et_al.db -T 10
The output from the anvi-run-hmms step showed 0 rRNA gene hits, which was in agreement with the authors’ statement in the paper. But the same output indicated that there were some bacterial single-copy core genes in the assembly.
Whenever there is a new contigs database, I take a quick look at the occurrence of the number of bacterial and archaeal single-copy core genes, since this is a great way to roughly estimate the number of genomes one should expect to find in an assembly.
Running these two commands,
anvi-script-gen_stats_for_single_copy_genes.py Kowarsky_et_al.db
anvi-script-gen_stats_for_single_copy_genes.R Kowarsky_et_al.db.hits Kowarsky_et_al.db.genes
tells me that the assembly does not contain any highly complete bacterial genomes:
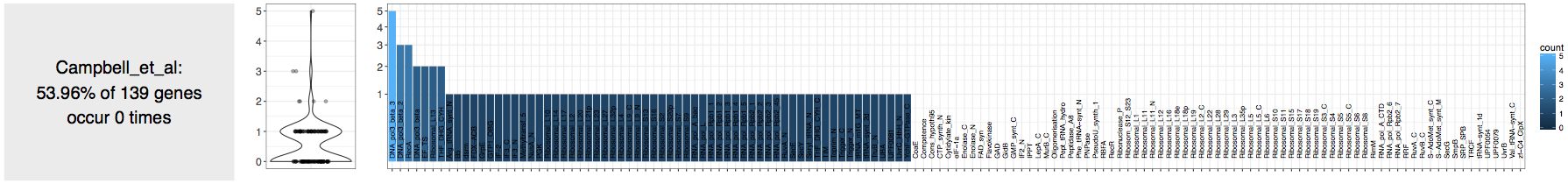
Fine.
But a closer inspection via thr good’ol squinting the eyes technique, reveals the occurrence of a good number of Ribosomal proteins in these contigs (which is also pointed out by the authors in their paper).
Sequences of ribosomal proteins
Next, I ask anvi’o to give me the amino acid sequences of all ribosomal proteins:
# first list all the available gene names in Campbell et al.
# single-copy core gene collection (the output is not shown):
anvi-get-sequences-for-hmm-hits -c Kowarsky_et_al.db \
--hmm-source Campbell_et_al \
--list-available-gene-names
# and from there I select all the ribosomal genes, and ask
# for their amino acid seqeunces to be stored in
anvi-get-sequences-for-hmm-hits -c Kowarsky_et_al.db \
--hmm-source Campbell_et_al \
-o Kowarsky_et_al_SCG_hits.fa \
--get-aa-sequences \
--gene-names "Ribosom_S12_S23, Ribosomal_L1, Ribosomal_L10, Ribosomal_L11, Ribosomal_L11_N, \
Ribosomal_L12, Ribosomal_L13, Ribosomal_L14, Ribosomal_L16, Ribosomal_L17, \
Ribosomal_L18e, Ribosomal_L18p, Ribosomal_L19, Ribosomal_L2, Ribosomal_L20, \
Ribosomal_L21p, Ribosomal_L22, Ribosomal_L23, Ribosomal_L27, Ribosomal_L28, \
Ribosomal_L29, Ribosomal_L2_C, Ribosomal_L3, Ribosomal_L32p, Ribosomal_L35p, \
Ribosomal_L4, Ribosomal_L5, Ribosomal_L5_C, Ribosomal_L6, Ribosomal_L9_C, \
Ribosomal_L9_N, Ribosomal_S10, Ribosomal_S11, Ribosomal_S13, Ribosomal_S15, \
Ribosomal_S16, Ribosomal_S17, Ribosomal_S18, Ribosomal_S19, Ribosomal_S2, \
Ribosomal_S20p, Ribosomal_S3_C, Ribosomal_S4, Ribosomal_S5, Ribosomal_S5_C, \
Ribosomal_S6, Ribosomal_S7"
The resulting FASTA file is here. But I summarized the output for you a tiny bit:
| Found in the assembly | e-value | Contig | Hit start | Hit stop |
|---|---|---|---|---|
| Ribosomal_L20 | 5.5e-36 | PR_node_48 | 6269 | 6617 |
| Ribosomal_L32p | 1.1e-09 | PR_node_60 | 1388 | 1538 |
| Ribosomal_L9_N | 4.7e-18 | PR_node_197 | 1959 | 2403 |
| Ribosomal_L27 | 7.3e-34 | PR_node_197 | 2470 | 2749 |
| Ribosomal_S16 | 8.7e-15 | PR_node_199 | 1751 | 2105 |
| Ribosomal_L13 | 1.4e-33 | PR_node_260 | 1670 | 2027 |
| Ribosomal_L17 | 1.9e-36 | PR_node_260 | 2032 | 2383 |
| Ribosomal_L13 | 7.3e-32 | PR_node_287 | 1680 | 2406 |
| Ribosomal_S2 | 3.8e-62 | PR_node_296 | 1014 | 1740 |
| Ribosomal_L21p | 4.2e-28 | PR_node_340 | 1693 | 2065 |
| Ribosomal_L2 | 1.1e-28 | PR_node_444 | 1 | 280 |
| Ribosomal_L23 | 1.3e-21 | PR_node_444 | 325 | 619 |
| Ribosomal_L4 | 1.1e-60 | PR_node_444 | 628 | 1270 |
| Ribosomal_S7 | 8.3e-56 | PR_node_1196 | 0 | 477 |
| Ribosomal_S20p | 7.7e-15 | PR_node_1285 | 146 | 410 |
| Ribosomal_L10 | 8.6e-23 | PR_node_1290 | 148 | 637 |
| Ribosomal_L14 | 5.1e-30 | HT_node_2492 | 11 | 407 |
| Ribosomal_S13 | 2.9e-25 | LT_node_1007 | 1 | 250 |
Authors mention that they found ribosomal proteins in 14 contigs, which perfectly matches to the table above: there are 14 distinct contigs that carry ribosomal proteins in my results as well (it is always a great feeling to find perfect agreement with published material).
As you can see from this output, multiple of these hits are occurring in the same contig (i.e., PR_node_260 contains two ribosomal proteins one after another, etc), however, we don’t know whether any of these distinct contigs are coming from the same population.
The supplementary methods suggest that these contigs are assembled independently from each dataset. As a final note on this, in Kowarsky et al.’s notation in the study PR, HT, and LT stand for samples originate from ‘pregnant’, ‘heart transplant’, and ‘lung transplant’ individuals.
So one can say most of the contigs with ribosomal proteins are coming from blood samples of pregnant individuals.
Fine.
Since there isn’t much information on the taxonomic origins of these bacterial hits in Kowarsky et al.’s study, I decided to focus on these ribosomal proteins a bit more.
Searching ribosomal proteins in NCBI’s nr database
I simply went to the NCBI and used blastp to search those amino acid seqeunces in the database of non-redundant protein sequences.
Once the search was done, I clicked the Download menu, and selected Multiple-file JSON option.
I had never tried to make sense of JSON files form NCBI BLAST output before, so I quickly (and dirtily) wrote the following Python program to get the best hit for each query sequence:
#!/usr/bin/env python
import sys
import json
import glob
# poor man's whatever:
QUERY = lambda: hits['BlastOutput2']['report']['results']['search']['query_title'].split('___')[0]
QLEN = lambda: hits['BlastOutput2']['report']['results']['search']['query_len']
HIT = lambda: hits['BlastOutput2']['report']['results']['search']['hits'][index]
DESC = lambda: HIT()['description'][index]
TITLE = lambda: DESC()['title']
SCINAME = lambda: DESC()['sciname']
ACC = lambda: '[%(desc)s](https://www.ncbi.nlm.nih.gov/protein/%(desc)s)' % {'desc': DESC()['accession']}
HSPS = lambda: HIT()['hsps'][0]
PCTALIGN = lambda: str(HSPS()['align_len']* 100 / QLEN()) + '%'
PCTID = lambda: str(HSPS()['identity'] * 100 / HSPS()['align_len']) + '%'
print('|'.join(['', 'Found in the assembly', 'Best hit on NCBI', 'Percent alignment', 'Percent identity', 'Accession', '']))
print('|'.join(['', ':--', ':--', ':--:', ':--:', ':--:', '']))
# go through every json file in the directory:
for j in glob.glob('*.json'):
hits = json.load(open(j))
# skip the poop file
if 'BlastOutput2' not in hits:
continue
# report the best hit:
index = 0
# unless the best hit resolves to a multispecies .. if it does, increment
# index
while 1:
if TITLE().find('MULTISPECIES') == -1:
break
index += 1
print('|'.join(['', QUERY(), SCINAME(), PCTALIGN(), PCTID(), ACC(), '']))
When it is run in a directory of JSON files downloaded form the NCBI, this program produces a markdown-formatted output table, so lazy people like myself can copy-paste it on their blogs:
| Found in the assembly | Best hit on NCBI | Percent alignment | Percent identity | Accession |
|---|---|---|---|---|
| Ribosomal_S2 | Candidatus Taylorbacteria bacterium RIFCSPHIGHO2_01_FULL_46_22b | 96% | 58% | OHA18677 |
| Ribosomal_L21p | Parcubacteria (Nomurabacteria) bacterium GW2011_GWD2_39_12 | 83% | 58% | KKR01970 |
| Ribosomal_L2 | Parcubacteria (Nomurabacteria) bacterium GW2011_GWE2_36_115 | 100% | 79% | KKP91491 |
| Ribosomal_L23 | Candidatus Zambryskibacteria bacterium RIFCSPHIGHO2_02_FULL_38_22 | 97% | 52% | OHA94501 |
| Ribosomal_L4 | Candidatus Lloydbacteria bacterium RIFOXYC12_FULL_46_25 | 95% | 63% | OGZ17743 |
| Ribosomal_S7 | Candidatus Zambryskibacteria bacterium RIFCSPHIGHO2_01_FULL_39_63 | 98% | 72% | OHA86974 |
| Ribosomal_S20p | Parcubacteria bacterium GW2011_GWA2_47_12 | 97% | 61% | KKU59139 |
| Ribosomal_L10 | Candidatus Zambryskibacteria bacterium RIFCSPHIGHO2_02_38_10.5 | 99% | 60% | OHA92782 |
| Ribosomal_L20 | Candidatus Zambryskibacteria bacterium RIFCSPHIGHO2_01_FULL_43_27 | 96% | 64% | OHA89065 |
| Ribosomal_L32p | Candidatus Lloydbacteria bacterium RIFCSPHIGHO2_01_FULL_41_20 | 95% | 44% | OGZ03679 |
| Ribosomal_L9_N | Parcubacteria bacterium GW2011_GWA2_49_9 | 99% | 55% | KKW11623 |
| Ribosomal_L27 | Parcubacteria (Nomurabacteria) bacterium GW2011_GWF1_34_20 | 96% | 71% | KKP60887 |
| Ribosomal_S16 | Parcubacteria (Nomurabacteria) bacterium GW2011_GWF2_36_126 | 91% | 65% | KKP93401 |
| Ribosomal_L13 | Candidatus Zambryskibacteria bacterium RIFCSPLOWO2_12_FULL_39_23 | 97% | 66% | OHB12744 |
| Ribosomal_L17 | Candidatus Lloydbacteria bacterium RIFCSPHIGHO2_01_FULL_41_20 | 100% | 64% | OGZ03809 |
| Ribosomal_L13 | Polaromonas naphthalenivorans CJ2 | 38% | 96% | A1VJJ3 |
| Ribosomal_L14 | Rhodospirillales bacterium 69-11 | 81% | 36% | OJW27693 |
| Ribosomal_S13 | Enterococcus faecalis TX2141 | 100% | 100% | EFT89376 |
This is a bit crazy.
Amino acid sequences of these ribosomal proteins have reasaonable hits in the NCBI, and most of these hits are most closely related to Candidate Phyla Radiation genomes released in Christopher T. Brown et al.’s 2015 paper and Karthik Anantharaman et al.’s recent publication. Another interesting thing is that except the last two lines in this table, all these genes are coming from contigs found in assemblies of pregnant samples.
Some speculations
Before making any suggestions, there are two points that we should take into consideration.
Is it a single population?
The first point is that all these contigs may be coming from a single microbial population.
In fact, in my opinion, it is very likely since there are only a small number of ribosomal proteins that occur only once.
To test this I did something very quick and dirty with anvi’o. You know, using some very basic characteristics of metagenomic data, we can get genomes from metagenomes rather rapidly. However, these methods work best when ‘coverage’ data is available (see this for why). But even with sequence composition alone, we can see the emergence of bins. Using only this contigs database, I created a blank profile with anvi’o:
anvi-profile -c Kowarsky_et_al.db \
-o PROFILE \
-S Kowarsky_et_al \
--blank
Then I created a simple text file to identify contigs with ribosomal protein hits, as well as contigs that originate from the assemblies of pregnant data sets. Here is the file, and one could download it into their work directory this way:
curl -L -O http://merenlab.org/files/Kowarsky_et_al_additional_data.txt
Then I run the anvi-interactive on these:
anvi-interactive -c Kowarsky_et_al.db \
-p PROFILE/PROFILE.db \
--additional-layers Kowarsky_et_al_additional_data.txt
When I hit the draw button, this is what I had in my browser:
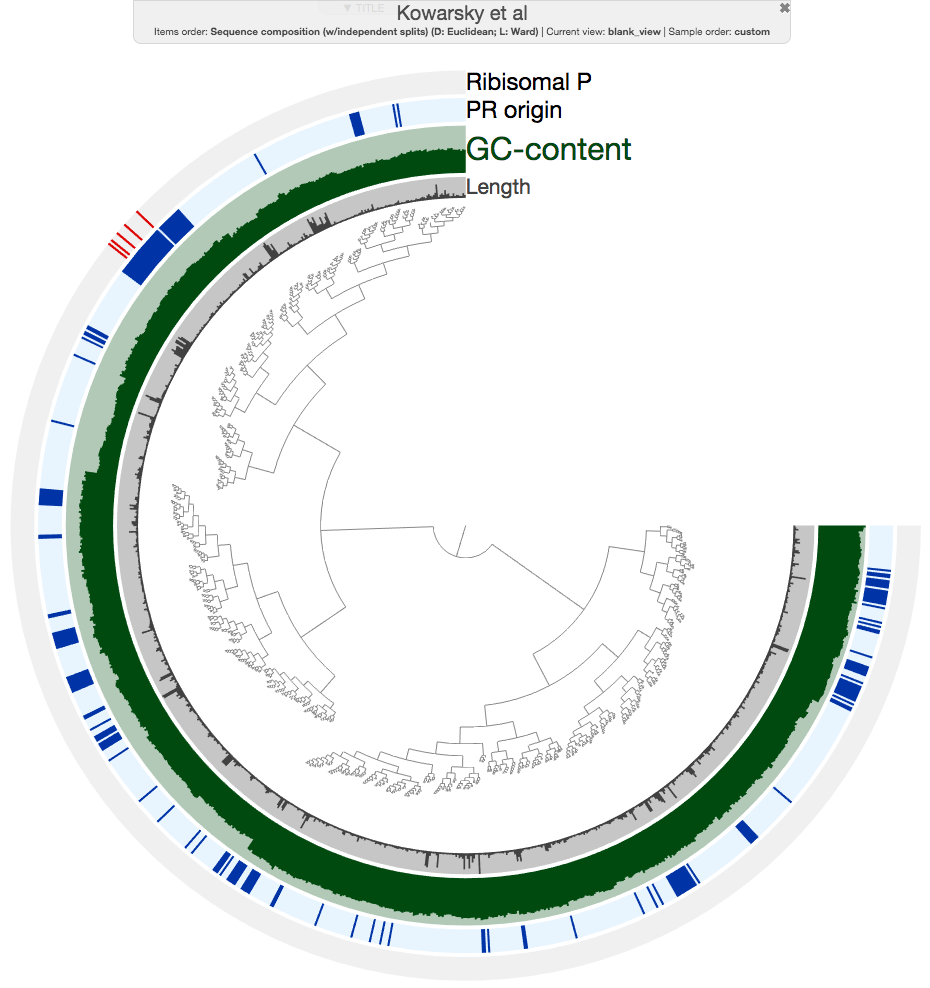
The ‘Ribosomal P’ layer marks contigs with ribosomal protein hits, and ‘PR origin’ layer indicates marks contigs from blood samples from pregnant women.
As you can see, all contigs with ribosomal protein hits are nicely together in a rather good looking cluster. But more importantly, there is a group of contigs that overlap with that section that are identified as ‘PR origin’ (the ones in closer brances are all from heart transplant, and lung transplant samples). So I made a very conservative selection here to minimize contamination:
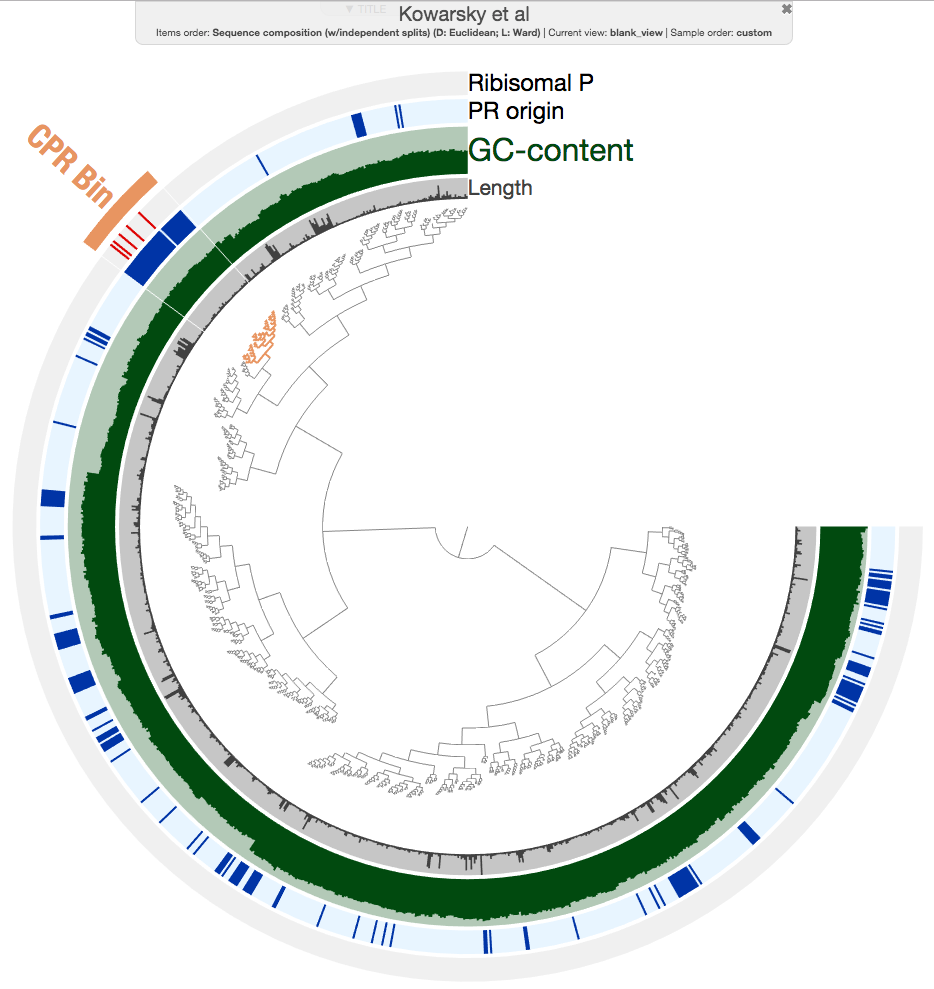
This bin is only 20% complete according to the bacterial single-copy core genes from Campbell et al.:
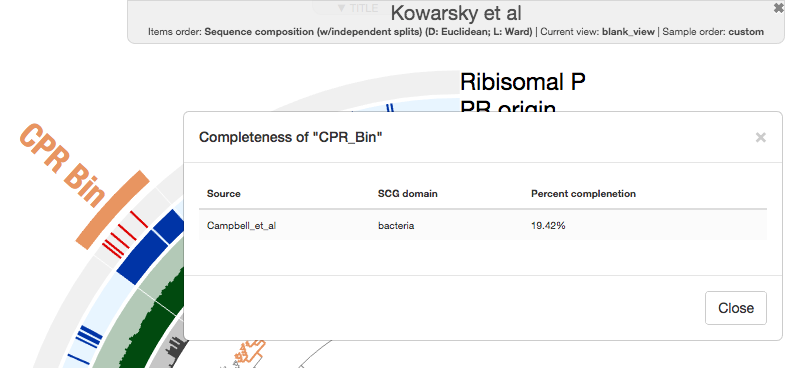
It is about 160K in length (about 1/5th of an average CPR genome), but it is infinite times larger than zero, and it probably has very minimal contamination if any. So far so good!
So at this point I am convinced this is a single population, and this is the anvi’o-reported FASTA file for it if you would like to play with it more: CPR_Bin-from-Kowarsky-et-al.fa
Remember that this bin is recovered from very weak data, and it is very likely contains some noisy bits and pieces despite best efforts.
Is it in a single individual?
The second point is about its prevalence, and the quick answer is “we don’t know because Meren is lazy“.
Just like the fact that this seems to be a single population in the dataset, it may be occurring in a single individual. Becasue CPR genomes are often quite lonley in the sequence space, in our experience, they get assembled very nicely even in very complex samples, or even when they are not quite abundant (i.e., our recovery of CPRs form TARA Oceans metagenomes). The next very important step is to map all metagenomes to this bin to see whether all contigs in it recruits reads from multiple individuals. If you would like to do that, you can get all metagenomic short reads from ‘pregnant’ data sets, and map those to the contigs in this file.
If there is a single individual, it may be possible for someone to go back to that sample, and sequence it deeper to recover a good CPR genome bin.
Hypothesis: Oral cavity may be the actual reservoir for this CPR (Concluision: Not likely)
A very reasonable hypothesis came from Clifford Beall on Twitter after this post appeared online: “Blood bacteria sometimes come from the oral cavity and some CPR are there, could be one of those”.
In our -limited- experience in our lab, CPRs we find in the oral cavity consistently hits to TM7 (a.k.a Saccharibacteria) with rather high identities. However, sequence identities are too low for genes in this bin compared to what we had been seeing. That being said, probably if we understand its prevalence (which requires some mapping), we could have a better idea about its origin. The next thing to do is to map some oral metagenomes to this bin. If you do that, feel free to send me the BAM files or let me know about your findings!
Clifford Beall reports back: no hits from 20 saliva metagenomes
Clifford Beall reported on Twitter that his mapping of 20 saliva metagenomes with 68M paired-end reads didn’t yield any mapping that looked real to him.
Meren confirms: no hits from 14 plaque and tongue metagenomes
So, I also tried to recruit read using this bin from our plaque and tongue metagenomes that contains 587 million quality filtered reads in total. From these half a billion reads, the CPR bin recruited one read. Seriously. One as in one. Since it is only a single read, I will just put it there:
GCTCCGTCCATTTGAGCGGCACCAGTGATCATGTTTTTAACGTAGTCCGCGTGTCCTGGAGCGTCGATGTGAGCGTAGTGACG
The lucky contig is the 4,442 nt long PR_node_96. This contig does not really have a good match on the NCBI, yet the read from the metagenome matches 100% to multple Streptococcus isolates on the NCBI, including usual suspects of oral cavity like S. gordonii, S. mitis, S. sanguins. But the read does not match so perfectly to the contig (but Bowtie2 is so awesome, it managed to find it for us):
Score Expect Identities Gaps Strand
109 bits(120) 3e-28 74/83(89%) 0/83(0%) Plus/Plus
Query 1 GCTCCGTCCATTTGAGCGGCACCAGTGATCATGTTTTTAACGTAGTCCGCGTGTCCTGGA 60
|| |||||||| ||||||||||||||||||||||| || | ||||| ||||| ||||||
PR_96 4342 GCACCGTCCATCTGAGCGGCACCAGTGATCATGTTCTTGATATAGTCAGCGTGACCTGGA 4401
Query 61 GCGTCGATGTGAGCGTAGTGACG 83
|||||||||||||||||||| ||
PR_96 4402 GCGTCGATGTGAGCGTAGTGGCG 4424
I was so surprised by the fact that only one out of half a billion reads were recruited, I started to doubt that there may have been a mistake with the workflow.
So, to test the workflow, I downloaded a random isolate genome that I know occurs a lot in the oral cavity (which happened to be Streptococcus mitis B6 FN568063.1), and used the same exact workflow to map the same metagenomes to that genome, instead of the CPR bin. Instead of one, S. mitis recruited 687,973 reads. So, the workflow seems to be working well, but this CPR bin (1) is nowhere to be found in our oral metagenomes, and (2) does not carry any genes that have any reasonable resemblance to anything in our oral metagenomes.
I’m a bit more curious now.
Functions
Since this is a tiny, incomplete CPR bin with only 203 genes, it is clear that it would be very unlikely to be able to make any conclusive statements regarding its role. But I thought it still wouldn’t hurt to take a look at its functions (since it is very easy with anvi’o (shameless plugs all over)).
I first run NCBI COGs on it:
anvi-run-ncbi-cogs -c Kowarsky_CPR-contigs.db \
--num-threads 10
And created a summary (from which I could get all the gene sequences and functions for the CPR bin):
anvi-summarize -c Kowarsky_CPR-contigs.db \
-p PROFILE/PROFILE.db \
-C default \
-o SUMMARY
From that output, this is the most important file that contains all gene calls and COG functions in the CPR bin: CPR_Bin-from-Kowarsky-et-al-functions.txt
OK.
I myself gazed through the functions very quickly and recognized some that are common to many bacterial genomes, such as histidine kinases, tRNA synthetases, cell division proteins, ABC transporters, etc. Good that this thing really looks like a bacterial genome. One of the first thing that caught my eye because I didn’t recognize them quickly were transketolase genes.
Besides multiple genes for transketolase subunits, one of the contigs carried in a row genes for Transketolase, Pentose-5-phosphate-3-epimerase, Ribose 5-phosphate isomerase, and Glyceraldehyde-3-phosphate dehydrogenase. When my Google search for ‘transketolase’ returned these two papers in the first page, I started to think that someone might be playing a trick on me so I waste weeks of my life on something I know very little about:
OK. In all seriousness, transketolase turns out to be an “important enzyme in the breakdown of glucose through the pentose phosphate pathway”. For instance, in mammals, transketolase connects the pentose phosphate pathway to glycolysis. And the pentose phosphate pathway is a “metabolic pathway parallel to glycolysis” (here is a very nice introductory lecture on this).
The occurrence of these genes in such a lovely synteny suggests that this bin has some sort of pentose phosphate pathway, and it can use products of glycolysis towards the generation of nicotinamide adenine dinucleotide phosphate (NADPH). NADPH is an essential electron donor for anabolic reactions in all domains of life, so the pentose phosphate pathway is pretty common among microbes. To confirm that, I search for COG0036 (Pentose-5-phosphate-3-epimerase) in my collection of 4,233 gold-standard bacterial and archaeal genomes I got from Ensembl, and in fact 43% of all genomes had at least one gene that resolved to this COG. So yeah, not that interesting so far.
But in comparison, of all genomes, 28% have COG0698 (Ribose 5-phosphate isomerase), and 12% have COG3959 (Transketolase, N-terminal subunit). And only 9% of all genomes had all of them together. So even though the pentose phosphate pathway is very common, it seems the one that uses the enzyme transketolase to connect the pentose phosphate pathway to glycolysis is not that common. But is there something common to all those not-that-common geomes? The genomes that had the pentose phosphate pathway genes with transketolase included these guys: Atopobium parvulum, Bacillus licheniformis, Bacteroides vulgatus, Escherichia coli, Klebsiella pneumoniae, Lactobacillus gasseri, Listeria monocytogenes, Mycobacterium smegmatis, Salmonella enterica, Streptococcus pneumoniae, and Yersinia pestis. Most of these are either well-known human pathogens found in blood, or human associated microbes. In fact, the ones that belonged to phyla Proteobacteria, Firmicutes, and Bacteroidetes, made up 77% of all 9% of genomes that had this pathway similar to the one in this CPR bin.
So. Is it safe to say there is something up? Can we say this CPR has some metabolic capabilities that are similar to those pathogens that know how to find their ways in blood? There are number of studies that show the reduced metabolic capabilties and parasitic nature of some of the CPR genomes. So, if I had no shame, I would have insinuated that this CPR bin may suggest the existence of parasitic bacterial populations that can live in mamallian blood by relying on excess glucose in blood. As you know, even if this was true we may have not known about them as they could have been eluding cultivation and marker gene surveys due to their reduced metabolisms and quirky ribosomal proteins just like most other members of the CPR.
I think at this point it would be best to identify the patient these contigs assembled from, and do a much deeper seqeuncing of that sample if it is still available. Meanwhile, mapping publicly available blood metagenomes, or designing PCR primers based on this bin to serach for evidence for its occurrence in other individuals may be potential directions if there is any interest.
Can we say it though?
Regardless of all these, (1) assuming that nothing went wrong with the sampling, library preparation, and sequencing procedures, and (2) after taking into consideration that we may be talking about a single microbial population observed in a single pregnant woman, and (3) after reminding ourselves that this population may not even be active and simply just sweeping into the blood stream from the oral cavity of an individual, it still is probably safe to say for the first time “CPR in human blood!”.
There you have it ;)
New blog post. Investigating some of the microbial contigs assembled from human blood metagenomes by Kowarsky et al. https://t.co/9wd60xd1nh
— A. Murat Eren (@merenbey) August 24, 2017