

Table of Contents
- Point #1: Emulsion PCR is bad
- Point #2: DWH O. desum is low quality and not that abundant in the oil plume
- Point #3: Disconnected 16S gene
- Point #4: DWH O. desum and ‘Ca B. macondoprimitus’ are similar anyway
- Point #5: blastx says ‘Ca B. macondoprimitus’ was there
- Point #6: Plume Oceanospirillaceae was diverse, ‘Ca B. macondoprimitus’ almost matched to a subset of it
- Point #7: DWH O. desum is very different than Mason et al. SAGs
- Conclusion: DWH O. desum does not represent the dominant Oceanospirillaceae population, while ‘Ca B. macondoprimitus’ does
TL;DR: There was this PNAS paper from Gary Andersen and his colleagues, which contained various claims that we did not agree with, so we wrote a letter:
which appeared in PNAS along with their response to our letter:
We thought the response, similar to the original study, contained claims that were not supported by data. But since there is no way to respond to these rebuttals through PNAS, we are doing it here. You know why? Because this is the age of internet, and journals no longer get to decide when the conversation ends.
The post contains some background into the story, the summary of our letter and the rebuttal, and finally our response to the rebuttal.
There is even a poem in it.
Introducing the oil plume debate: The four studies
Most of you who read these lines know about the Deepwater Horizon Oil Spill that resulted in an environmental disaster in the Gulf of Mexico.
The oil leaked from the deep-sea well BP failed to secure after the blowout, formed a plume with trapped oil and gas. Here is Jack Cook’s illustration to have a mental picture of this oil plume:
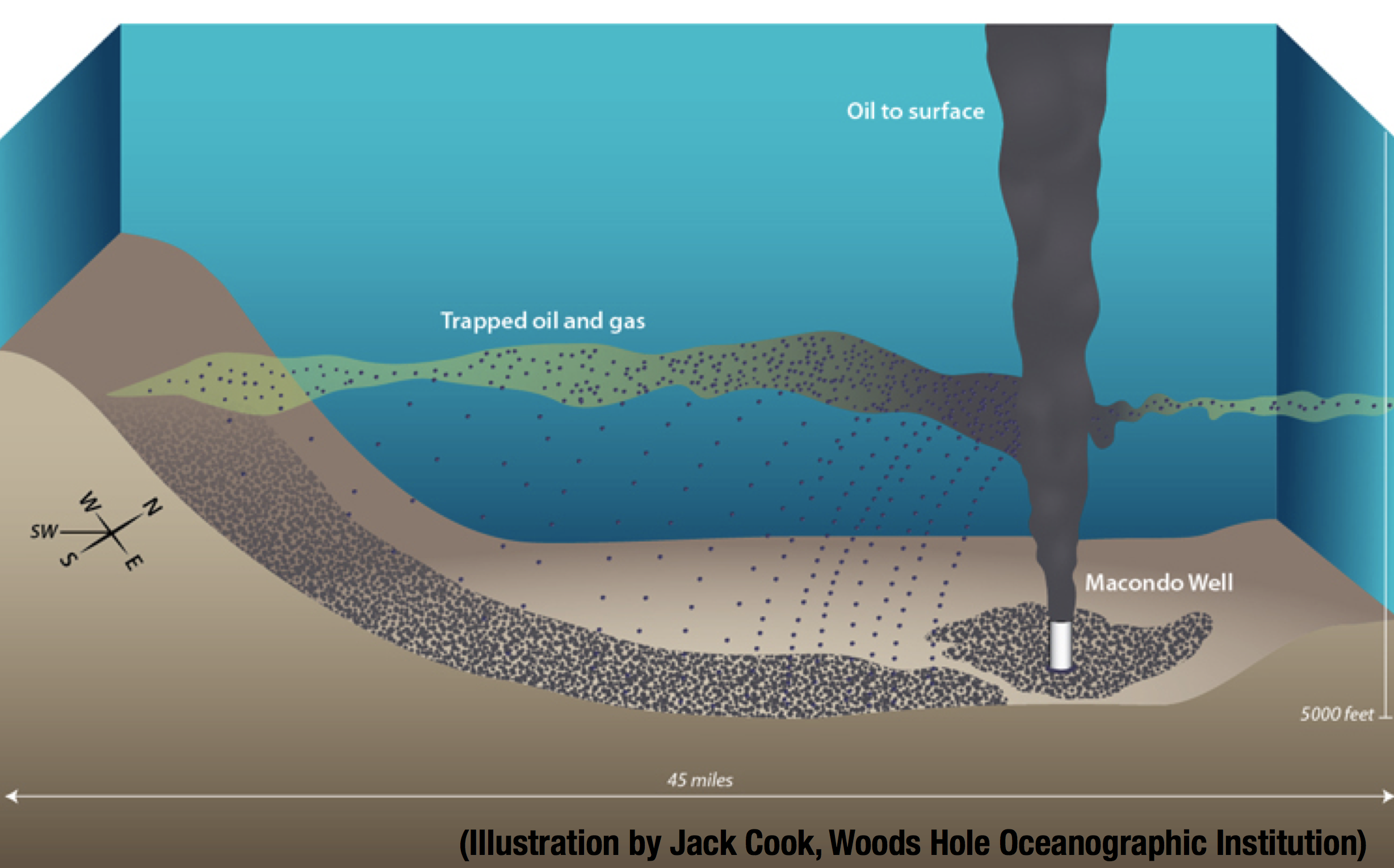
The oil plume created a lot of interest among microbial ecologists to better understand the functioning of the oil-degrading microbes that dominated it. The literature contains many studies related to this event, but here we will particularly mention four of those to set the stage for the current debate.
Study #1
The first study that provided some insights into the microbial makeup of the oil plume came from Hazen et al. (2010), in which the authors generated full-length 16S rRNA gene sequences from the oil plume using clone libraries and Sanger sequencing. This study revealed that a group affiliated to family Oceanospirillaceae dominated the plume, but not the uncontaminated areas surrounding the oil spill. This result was confirmed by many following studies, and it is now commonly accepted that the oil plume was initially dominated by an Oceanospirillaceae lineage. However, attempts to cultivate members of this lineage failed.
Study #2
The second study that is relevant to this debate is a follow-up study from Mason et al. (2012). The authors in this study generated metagenomes, metatranscriptomes, and single cell genomes (SAGs) from the oil plume samples. Metagenomes and metatranscriptomes were dominated by Oceanospirillaceae, consistent with results from Hazen et al. (2010). SAGs were also affiliated with Oceanospirillaceae, yet their 16S rRNA genes were only 95% similar to the ones generated by Hazen et al. (2010). Nevertheless, authors suggested that the SAGs corresponded to the Oceanospirillaceae population that dominated the plume soon after the release of oil. The substantial difference between the 16S rRNA gene found in the single cell genomes and the one that was recovered by Hazen et al. should have alerted any careful reviewer to ask for the metagenomes to be mapped onto to SAGs to investigate their abundance in the oil plume. Yet, this was not done until three years later, which brings us to the third study.
Study #3
The third study is from us: Eren et al. (2015). The only reason we are a part of this debate is because in 2015 we were looking around to find a proper dataset to demonstrate how anvi’o could be used to combine metagenomes and metatranscriptomes. Mason et al.’s oil plume dataset was an excellent choice for this, and we had no clue what we were getting ourselves into. In our study we mapped reads from the oil plume metagenomes and metatranscriptomes onto the SAGs and found that most of their genomic content remained undetected. Thus, both 16S rRNA gene comparisons and genome-wide read recruitment results suggested that the SAGs did not represent the abundant Oceanospirillaceae lineage. In parallel, we recovered a partial population genome, DWH O. desum, by co-assembling the oil plume metagenomes. DWH O. desum has a length of 1.07 Mbp, it recruited more than 99% of all transcripts that mapped to the assembly from the sample that was closest to the well head, and a phylogenomic analysis of its single-copy core genes using CheckM linked it to Oceanospirillaceae. More strikingly, the 16S rRNA gene appeared in DWH O. desum bin was 100% identical to a dominant 16S rRNA gene generated by Hazen et al. (2010). Later, a phylogenomic analysis of oil degrading bacteria that included DWH O. desum by Brett Baker and his colleagues showed that DWH O. desum nicely sits together with other oil degrading bacteria. Given the taxonomic affiliation, 16S rRNA gene match, tremendous recruitment of metagenomic as well as metatranscriptomic reads that were not used for assembly or binning, we suggested that DWH O. desum provided access to the genomic context of an Oceanospirillaceae population that dominated the plume soon after the oil spill.
“Desum” means “missed” in Latin. Tom suggested to call the population genome tentatively with this name since Mason et al. (2012) could have reconstructed it years earlier. That said, we are very thankful to Olivia Mason, Janet Jansson, and their collaborators for providing the science community with such invaluable data from the oil plume, without which much of these debates would have been impossible.
Study #4
The fourth study comes from Gary Andersen and his colleagues. In the Hu et al. (2017) paper in PNAS they perform an incubation experiment in an attempt to recapitulate the environmental conditions that took place in the oil plume, and they successfully reconstruct a population genome that is affiliated with the family Oceanospirillaceae, which they call Bermanella macondoprimitus. ‘Ca B. macondoprimitus’ is a 2.54 Mbp near-complete genome, and in the study authors claim that it represents “the genome of the dominant hydrocarbon-degrading organism that was detected in the initial stage of the DWH plume”.
So, that’s that.
The letter, and the rebuttal
The recovery of a more complete genome of the most abundant early responder in the oil plume through a simulation experiment is a very exciting claim in many ways. However, we realized that, similar to SAGs in the Mason et al. study, the 16S rRNA gene in this population genome did not perfectly match any of the 16S rRNA genes generated by Hazen et al. (2010), and although they recruited reads from metagenomes of the simulation (Figure 3 in the Hu et al. study), the authors did not recruit reads from the oil plume metagenomes to support their claim about the environmental relevance of the population they found in their simulation. Finally, although they used DWH O. desum in their phylogenomic analysis (of course, after kindly renaming it to the taxon name of their liking), they did not use it to recruit reads from the simulation. Indeed using a population genome recovered from the oil plume for a recruitment analysis could provide the missing link between the environment and the simulation. Overall, they did not make good use of the metagenomes and metatranscriptomes that were generated from the environment they try to simulate, neither did they make a good use of the population genome that was recovered from the environment. Space considerations often limit the amount of analyses that can be fit into a single manuscript, however, (1) PNAS has a 10-page format (they opted for the 6-page format), and (2) no respectable journal refuses to make available supplementary information or data when space is the only limitation when authors wish to substantiate their claims. All these points intrigued use to take a more careful look at the study.
So. To see whether the ‘microbe mystery’ of the oil spill was really solved with this work as the Berkeley Labs claimed, we did what we though was missing from the study and used ‘Ca B. macondoprimitus’ to recruit reads from the metagenomes and metatranscriptomes generated from the oil plume to understand whether it was matching to populations found in the oil plume. We also used DWH O. desum to recruit reads from the metagenomes generated from the incubation experiment to understand whether it was enriched in this simulation.
We discovered that the large fraction of the genomic context of ‘Ca B. macondoprimitus’ was not detected in the environment. The same conclusion was true for DWH O. desum and the incubation experiment. Furthermore, the only oil degradation gene identified in Ca B. macondoprimitus, alkB did not recruit any reads neither from metagenomes nor transcriptomes, and we thought with heavy heart that we should let the community know that despite the importance of this study at gaining insights into the microbial succession after the initial response, it did not necessarily solve the ‘mystery’ of the environment. Which means there is more work to be done for everyone else to fully recapitulate the environmental conditions that took place during the oil spill. This is important, because failure to fully understand the dynamics behind microbial responses to similar situations may result in poor decisions at the policy making level.
Our letter made simple points based on these facts:
-
DWH O. desum was a genome reconstructed from the environment, ‘Ca B. macondoprimitus’ comes from an incubation experiment performed years later.
-
DWH O. desum had a 16S rRNA gene that is consistent with the results from Hazen et al. (2010), ‘Ca B. macondoprimitus’ did not.
-
DWH O. desum recruited a remarkable amount of reads from the oil plume metagenomes and metatranscriptomes, ‘Ca B. macondoprimitus’ did not.
Our letter showed that one of the main claims made in the fourth study did not hold given the environmental data, and they did not in fact enrich the dominant hydrocarbon-degrading organism that was detected in the initial stage of the DWH oil plume.
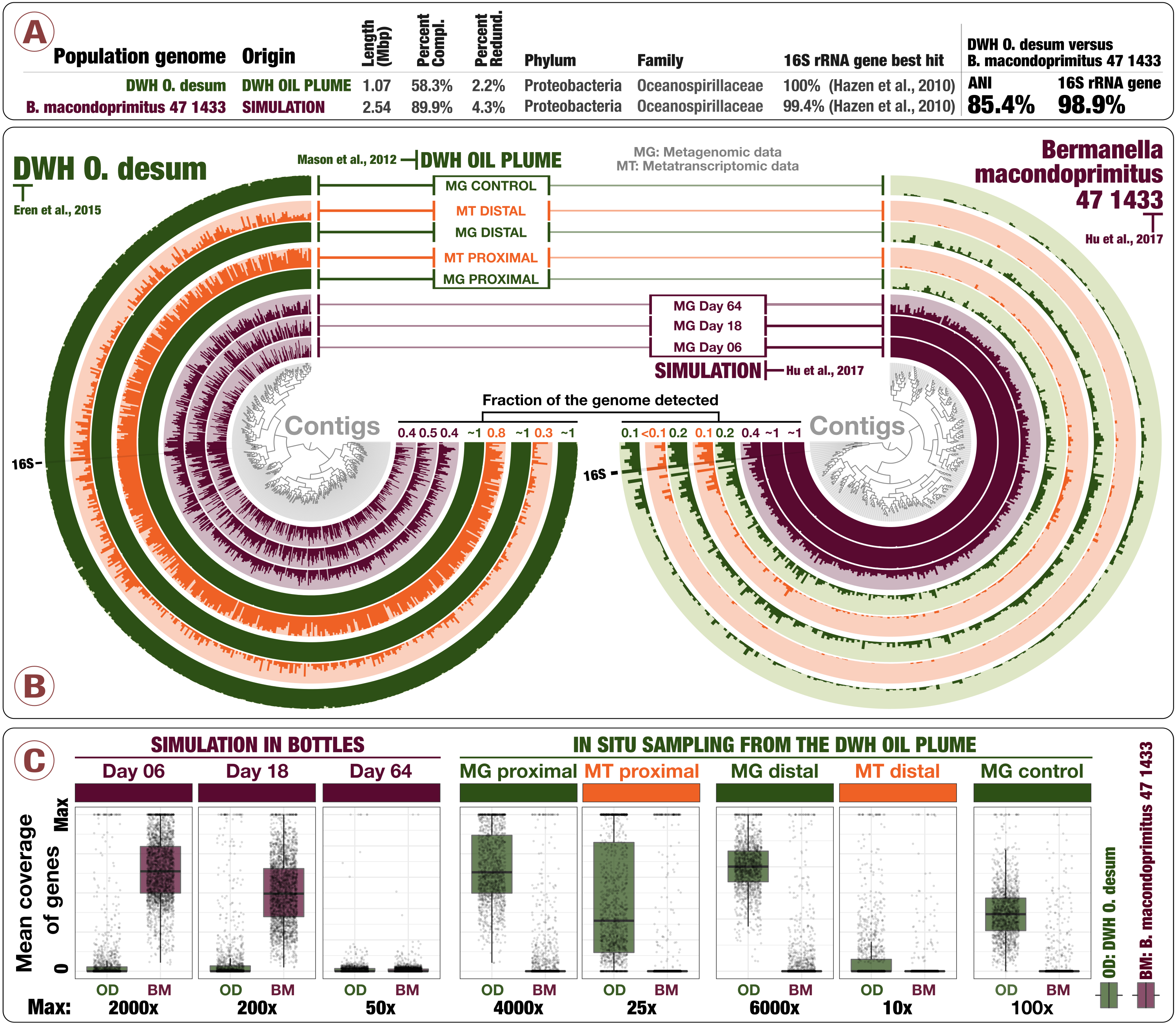
Our letter not only comes with a visual summary of it you see above, but also gives interactive access to the results for recruitment analyses for,
So you can right-click on any contig, and inspect their read recruitment at the nucleotide-level resolution yourself.
Our letter is now published:
The rebuttal from Gary Andersen and his colleagues to our letter is also here:
Our response to their rebuttal for our letter about their paper follows shortly, but first there is an important note we would like to make for the ones who still follow.
An important note
It is important to note that nothing in our letter suggests that ‘Ca B. macondoprimitus’ or the incubation experiment is irrelevant to the field of microbial ecology or has no value. In contrast, we believe they represent important research tools.
To be precise, our findings only suggest that there is no evidence to support the claim that ‘Ca B. macondoprimitus’ was abundant in the environment given all the environmental data. It may have been the case that ‘Ca B. macondoprimitus’ bloomed very early in the oil plume, and completely disappeared by the time Hazen et al. and Mason et al. finally made it to the event site for sampling after more than a month, missing the very early reasponders that may have included ‘Ca B. macondoprimitus’. But this is not what the Hu et al. paper suggested. Perhaps a more highly-resolved shotgun metagenomic survey of the incubation for the first 6-weeks period could have shed light on this.
It is up to the experts to conclude (1) whether ‘Ca B. macondoprimitus’ can be relevant to what was happening in the environment, and (2) to what extent a simulation that gave rise to a population genome significant parts of which was not detected in the environment could be used to make suggestions about the functioning of the environment.
Our response
In their rebuttal, Gary Andersen and his colleagues maintain their original claim, and make it clear that there is no reason to doubt that ‘Ca B. macondoprimitus’ represents the dominant Oceanospirillaceae population in the oil plume.
Their rebuttal ends with this sentence:
B. macondoprimitus population genome detected in the laboratory simulation represents a strain variant of the dominant Oceanospirillales that bloomed in deep oil plumes
You certainly have more important things to do, so here we offer an end to our response with a post-modern poem just so you can get back to work:
We get what we get, and we don't get upset.
While they are critical research tools,
it is not fair to expect them to be perfect,
because simulations are simulations,
and they can't beat the environment.
We may have already said a bit too much to not get upset,
and then it may be alright to be just a little sad,
but even then we shouldn't fight the data,
especially when it shows that,
our beloved alkB gene,
managed to recruit,
exactly zero reads,
from the environment.
There you have it. Now you can go. But if you are already too much into this, here are some of the points made in the rebuttal we thought we should address.
Point #1: Emulsion PCR is bad
In their response, the authors criticize the way the oil plume metagenomes were generated (which used emulsion PCR), suggesting that this process might have heavily distorted the observed abundance patterns:
Delmont and Eren drew their conclusions from methods that are critically flawed. Importantly, they used samples that underwent an emulsion PCR [4], (according to Mason et al. overloaded by ~20 molecules/droplet [5]), which likely resulted in a heavy distortion of the sequenced community and observed abundance patterns [6].
We agree that the emulsion PCR may bias coverage patterns. But the community composition of the oil plume metagenomes was consistent with those found by clone libraries of 16S rRNA genes published by Hazen et al. (2010), and this suggests that even though the abundance patterns may have been affected, its effect on the membership was likely negligible. The likely impact of the coverage bias on differential coverage-based binning is also negligible since there is only one abundant population in metagenomes from the plume, and anvi’o takes into consideration for each contig the abundance and tetranucleotide frequency statistics as well.
Furthermore, emulsion PCR alone can not explain the complete lack of read recruitment for most parts of the genome of ‘Ca B. macondoprimitus’, unless one tries very hard. For instance, trying very hard would require one to suggest that the emulsion PCR removed all DNA sequences matching to ‘Ca B. macondoprimitus’, but not the DNA sequences from other members of the Oceanospirillaceae lineage. In addition to that they would have to argue that the clone libraries by Hazen et al. also systematically missed the 16S rRNA gene of ‘Ca B. macondoprimitus’, but not from the others. Indeed there is a simpler, one-step explanation: ‘Ca B. macondoprimitus’ does not represent a member of the Oceanospirillaceae lineage that dominated the oil plume in the environment.
Point #2: DWH O. desum is low quality and not that abundant in the oil plume
The authors also emphasiz the higher quality of ‘Ca B. macondoprimitus’ compared to DWH O. desum:
Delmont and Eren failed to use any metagenome assembler that attempts to account for biases of amplified metagenomes and compounded the issue by applying a binning method that uses abundance information [6]. The quality of their recovered genomes is poor [6]: a genome of 1.6 Mbps without any single copy genes [6] and a highly fragmented and incomplete genome of the designated “dominant” organism called “O. desum” (Figure 1A/B).
It is correct that ‘Ca B. macondoprimitus’ is a high-quality, high-completion population genome, while the DWH O. desum published in 2015 is a highly fragmented, low-completion population genome. We got what we got, and we weren’t that upset. But then this information (1) has nothing to do with the fact that a significant amount of ‘Ca B. macondoprimitus’ fails to recruit any reads from the environment or that its 16S rRNA gene does not match any of the Hazen et al. sequences, and (2) does not provide any evidence to argue against our claim that ‘Ca B. macondoprimitus’ was not an important member of the early responding microbes to the oil plume.
Perhaps the purpose of this point was to demonstrate our ‘critically flawed’ methods authors gracefully mentioned. We are more ready than you can imagine to accept that we are the impostors with all the critically flawed methods, but the logic here is not really working that well: ‘Ca B. macondoprimitus’ is from an incubation experiment to mimic the oil plume, and DWH. O desum is from the oil plume itself. The rebuttal reads “B. macondoprimitus showed higher completeness (>99%) and substantially lower fragmentation despite its great abundance in the [simulation]”. While this statement is very correct, it is not relevant to any discussion regarding the superiority of alternative approaches.
But we took their suggestion seriously, and re-analyzed the oil plume metagenomes by performing new assemblies with metaSPAdes which was not available in 2015. We recovered a new population genome of DWH O. desum, except this time it is 2.7 Mbp long, 97.8% complete, 0% redundant. The Average Nucleotide Identity (ANI) is 99.59% between the new and the original DWH O. desum (which was only 1.07 Mbp long). The bad news for Gary Andersen and his colleagues is that the ANI between the new DWH O. desum population genome and ‘Ca B. macondoprimitus’ is still only 84.32% over less than 1 Mbp of alignment.
Please feel free to visit this post to read about the technical details of the recovery of “DWH O. desum v2”, or as we like to call it “Ca Nobermanella desum”, mostly because we are funny.
The authors also suggested that because DWH O. desum recruited only ~15% of the reads from the oil plume metagenomes, it likely did not represent the “abundant population”:
Notably, this “dominant” organism recruited only 15% of reads from the amplified metagenomic library of the proximal plume sample (stated as 77.8% of 19.7% of mapped reads, [6]).
If a population genome recruits only 15% of the reads from metagenomes, there may be other, more abundant populations. So this is a valid concern, but only if the population genome of concern is near-complete. DWH O. desum was only 1 Mbp. So one can in fact logic this one out: “if Oceanospirillaceae are around 3 Mbp, and if DWH O. desum, the partial population genome of 1 Mbp length, is recruiting 15% of all reads, the actual population it represents should be about 45% of the reads”.
Indeed, the new 97.8% complete population genome from the environment (which gives access to the same dominant environmental population we initally learned about through DWH O. desum) now recruits 46% to 61% of all short reads from the oil plume metagenomes, leaving much less room for arguments against its brutal dominance.
We couldn’t demonstrate this at this level of clarity only through the force of logic, and we are happy that we performed the new assembly. We are thankful for this exchange which pushed us to do that and strengthen this point. When logic is not available, we can always come to the rescue with our critically flawed methods.
Point #3: Disconnected 16S gene
Authors show that the 16S rRNA gene of ‘Ca B. macondoprimitus’ was more connected to the rest of the population genome compared to the one that is found in DWH O. desum:
While the 16S rRNA gene of B. macondoprimitus can be linked to its genome via sequence overlaps (Figure 1C), there is no evidence that the 16S rRNA gene attributed to “O. desum” is connected to its recovered genome (Figure 1D).
This is true, but this also does not contribute to our understanding of the abundance of ‘Ca B. macondoprimitus’ in the oil plume.
Besides, although the 16S rRNA gene sequence is not connected to the initial population genome of DWH O. desum, it is one of a small number of contigs that are consistently covered above 4,000X in the environmental metagenome. Given that there is only a single population in the metagenome that is covered at that level, the possibility of the 16S rRNA gene to belong to another population is extremely miniscule. To appreciate this point, please take a look at the Panel B of this figure, while taking into consideration that it displays the log-normalized recruitment statistics for each contig from the entire metagenome.
Point #4: DWH O. desum and ‘Ca B. macondoprimitus’ are similar anyway
To make this point, authors describe a 16S rRNA gene analysis, bringing the big guns to a metagenomics fight.
Detailed analysis of 16S rRNA genes from the Gulf of Mexico revealed a massive population heterogeneity of Oceanospirillales represented by 146 different genotypes in a single cluster (Figure 2A), which group with “O. desum” and B. macondoprimitus (Figure 2A).
In their sequence dissimilarity ordination, DWH O. desum and ‘Ca B. macondoprimitus’ appeared next to each other, along with “genotypes” from Hazen et al. (2010).
We hope it is clear to everyone involved in this discussion that the resolution of 16S rRNA genes are not suitable to accurately track individual populations across environments. This is the reason why we work with population genomes and we used genome-wide read recruitment to argue that ‘Ca B. macondoprimitus’ is not the population that dominated the oil spill as it was claimed by Hu et al. Even if ‘Ca B. macondoprimitus’ had a 16S rRNA gene that is identical to one of the Hazen et al. sequences, it wouldn’t have changed the fact that a large fraction of the genome recruited 0 reads from the environment (here the authors kindly cite the poem).
Point #5: blastx says ‘Ca B. macondoprimitus’ was there
So the authors perform a blastx analysis, and discover that 38% of metagenomic reads from an oil plume metagenomes had a match to ‘Ca B. macondoprimitus’ (bitscore >50):
A large fraction of reads (38%) from the amplified metagenome matched to B. macondoprimitus’ predicted proteome (blastx, bitscore >50).
This analysis is misleading due to its lack of stringency. A blastx analysis with bitscore >50 is much less stringent than read recruitment using Bowtie2 or BWA with their default parameters. Read recruitment gets us very close to track individual populations, while the BLAST analysis does not. This is exactly why a significant fraction of ‘Ca B. macondoprimitus’ clearly is not detected in the oil plume metagenomes using read recruitment, while blastx can match many reads to it. It would have been an insult to the authors if we assumed that they didn’t know this. But here is an attempt to put it in simple terms for those of you who may not be as experienced: ‘Ca B. macondoprimitus’ may have enough number of genes, the translated nucleotide sequences of which are just good enough to resemble other genes in the environment at a bitscore of 50 or less, but the claim “this is the population genome that represents the abundant oil degrading population of the environment” requires a very different kind of evidence .. a kind of evidence that does not exist given the data, and will not magically emerge while shuffling tools and playing with numbers.
Point #6: Plume Oceanospirillaceae was diverse, ‘Ca B. macondoprimitus’ almost matched to a subset of it
The authors also claim that many genotypes of the abundant Oceanospirillaceae population occurred in the oil plume, and that ‘Ca B. macondoprimitus’ almost matched to one of these genotypes:
Notably, both “O. desum” and B. macondoprimitus represent population genomes, and we discovered high 16S rRNA similarity (>99.99%) between strains in the simulation experiment and strains recovered from the oil plume (Figure 2B). (…) the datasets used by Delmont and Eren were generated from amplification of multiple genotypes of the same species, resulting in very fragmented genomes, and are ultimately not representative of the true population structure during the oil spill.
The analysis behind this point relies on the mismatches between ‘Ca B. macondoprimitus’ and Hazen et al. sequences.
To understand the complexity of the environment, we did something more conclusive, and have investigated single-nucleotide variants (SNVs) among the reads DWH O. desum recruited from metagenomes and metatranscriptomes of the oil plume.
Here is an example plot that shows the coverage of a random DWH O. Desum contig:
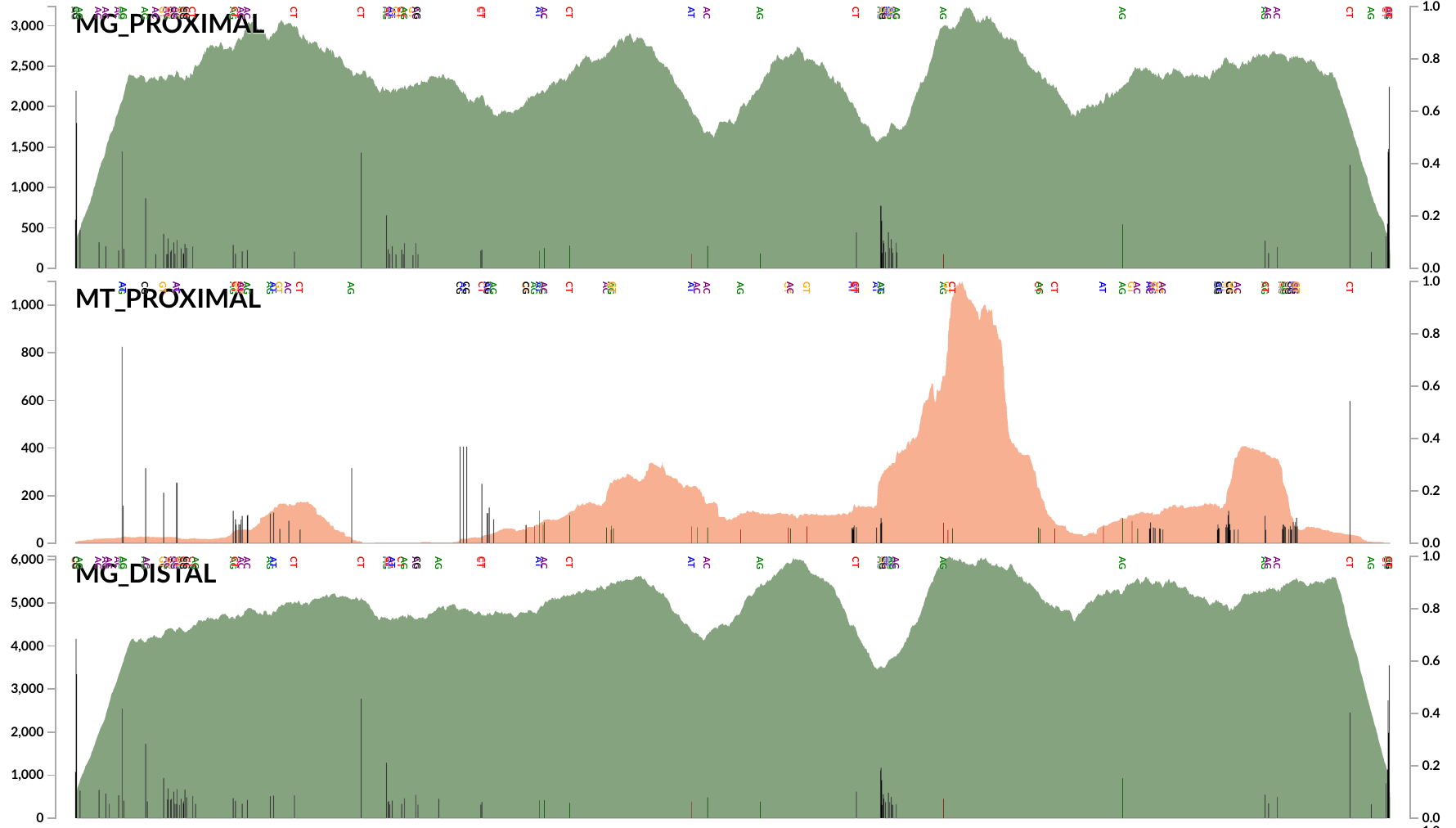
Every black bar in this coverage plot indicates the extent of disagreement (see the right-hand side Y-axis) between the environment and the genomic context.
The low-density of SNVs does not support the idea that “many genotypes of the abundant Oceanospirillaceae population occurred in the oil plume”. We probably are all on the same page that Hazen et al.’s 16S rRNA gene sequences and Mason et al.’s SAGs are great evidence to confidently suggest many populations of Oceanospirillaceae have occurred in the oil plume. But our analyses in our 2015 paper and here suggest that not all of them were abundant. In contrast, all publicly available data suggest that there was a single very abundant population by far, which was represented by the partial population genome DWH O. desum, and the SNV analysis above supports the idea that this Oceanospirillaceae cloud was not diverse enough to leave much room for other populations that may have been as abundant. Thanks to our critically flawed methods, these results are available here. Right clicking on any contig will allow one inspect the coverage and occurrence of SNVs across samples.
Ironically, this is a random contig from ‘Ca B. macondoprimitus’ (mapping results for which are also available here for your own investigation):
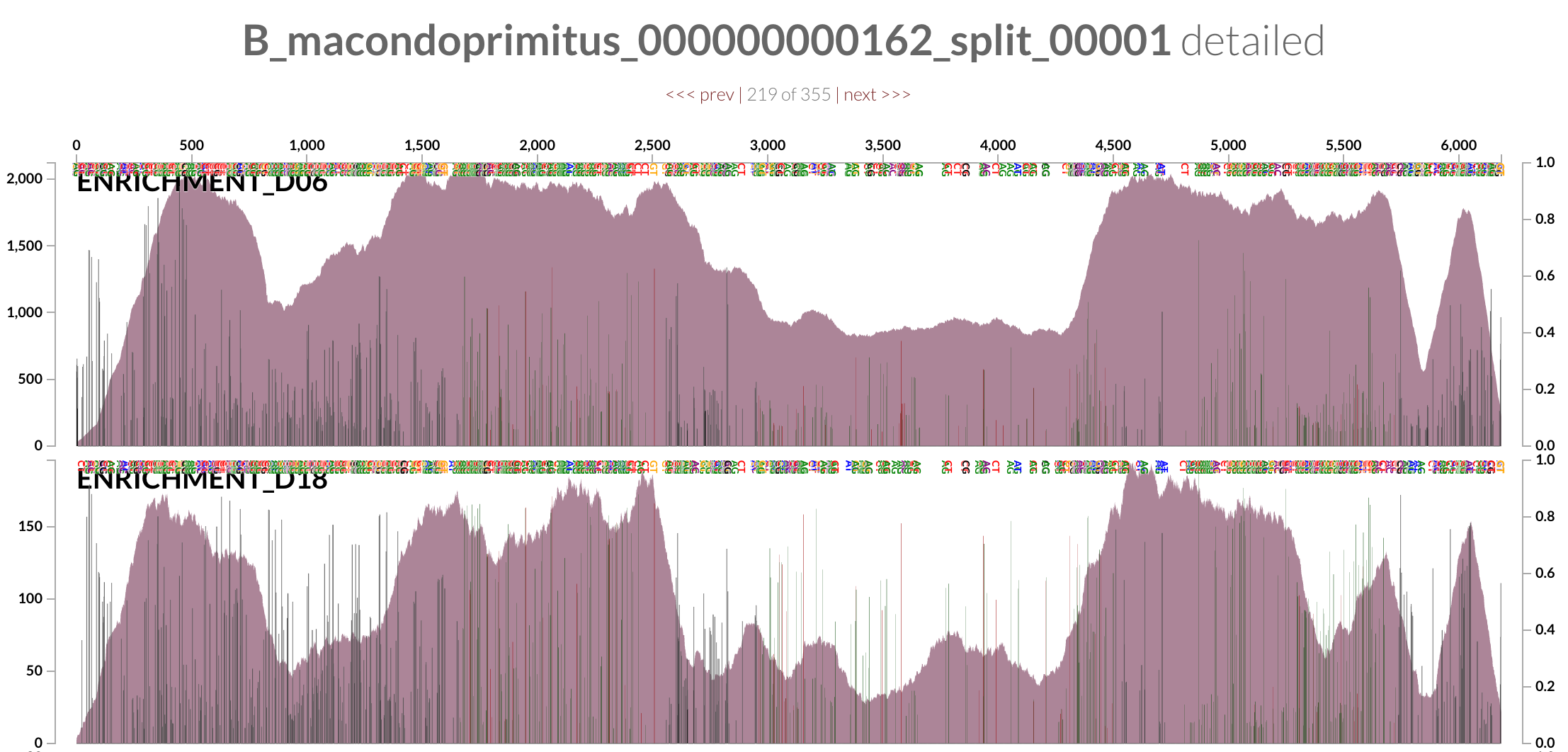
If we follow the logic laid out by the authors, the complexity of the metagenome from the incubation experiment would suggest that DWH O. desum is a better representative of the oil plume than ‘Ca B. macondoprimitus’ is of the incubation experiment (this is not the time, but it could be the topic of another discussion whether it is OK or not to put a species name on a genome that seems to be simply a consensus sequence for a highly diverse population).
Point #7: DWH O. desum is very different than Mason et al. SAGs
Authors also emphasized that DWH O. desum was very different from the SAGs generated by Mason et al. (2012):
Amplified metagenomes used by Delmont and Eren barely matched the Oceanospirillales single-cell genomes, although recovered from the same environment.
This was somewhat disappointing for us to read, because it showed us how little effort the authors put into understanding the point of our previous study, which contains an entire paragraph that explains how SAGs did not represent the abundant Oceanospirillaceae population, and remained undetected in the oil plume. Even the Probst et al. ordination in Figure 2A places the 16S rRNA gene sequences from these SAGs as distant from ‘Ca B. macondoprimitus’ as a random Bermanella isolate from the Red Sea, reiterating their irrelevance to the environment even when a low-resolution approach is used. So although it sounds like one, this is not a viable argument against DWH O. desum.
Conclusion: DWH O. desum does not represent the dominant Oceanospirillaceae population, while ‘Ca B. macondoprimitus’ does
The authors finally end their rebuttal restating that they have it:
(…) datasets used by Delmont and Eren were generated from amplification of multiple genotypes of the same species, resulting in very fragmented genomes, and are ultimately not representative of the true population structure during the oil spill. In contrast, the high-quality B. macondoprimitus population genome detected in our laboratory simulation represents a strain variant of the dominant Oceanospirillales that bloomed in deep-water oil plumes (…)
Certainly a nice way to finish a note that will not get an official answer, but given the data and the response, this does not sound anything more than wishful thinking.
‘Ca B. macondoprimitus’ was not a significant part of the oil plume. Do you want to see it yourself? Just click this link, and then inspect the contig that contains the alkB gene (it is the third left-most gene). You will first see that it is very well detected in the simulation, and if you continue scrolling down, you will also see that it recruits literally 0 reads from the environment.
In contrast, we now have a new near-complete population genome reconstructed from the oil plume metagenomes that recruits up to 61% of all metagenomic reads, and still has only 84.32% ANI over less than 1 Mbp of alignment to ‘Ca B. macondoprimitus’. Given the environmental data, ‘Ca B. macondoprimitus’ is no strain variant of anything that was abundant in the oil plume.
So, DWH O. Desum is the most covered genome in the environment. But ‘Ca B. macondoprimitus’ is the most covered genome in the media:
So everyone gets something at the end.
Meren’s 2 cents
For everyone who came this far to read this blog post I wanted to finish it with a rather positive note.
In case you haven’t had the chance to learn it yourself yet, I can tell you by experience that highlighting problems with published studies is not easy, especially when those studies you want to talk about include individuals you know personally or through the social media, interact with, read from, and respect in many ways. But maybe it is most important to continue raising concerns despite these social inconveniences, because every field of science is small enough that sooner or later we all end up knowing each other personally or through the social media, interacting, and finding reasons to respect each other. Although everyone would probably agree that criticism should not be sacrificed for social reasons, that ideal still does not make things any easier.
These hard-to-make criticisms take more than they give back. Re-analyzing data from others and writing about your concerns will often turn into a major time and energy sink as you will feel the crushing doubt about your accuracy. Then your findings will often largely be ignored. And at the very end of all maybe you will even realize that you are the Don Quixote of this story, fighting against something that everyone is already aware of, and chose to let go.
If you are lucky, you may randomly hear from someone at a conference or in an e-mail that they appreciated what you did. But if you expect anything more than that, such as a pat on the back or some collegial love, you better don’t even think about going that path. Meren, you said you were going to finish with a positive note :(
Well, although often times preparing or responding to criticism sucks and does not pay back much, other times things can work out rather nicely for the greater good. For instance, I find the fact that there is now a near-complete population genome from the oil plume itself as a very productive outcome of this particular discussion, and as something that is good for everyone. They also often turn into valuable opportunitues to improve things, and help outside observers to get exposed to most intricate details of methods and questions we all are struggling to better understand as a community.
Given how invested we all are into our science, it can also be hard to avoid occasional yelling competitions. But even they are OK in my opinion: science is complex, we all can miss or misunderstand subtle points, and get frustrated. But it is safe to assume that everyone is doing their best. This is what we get, and we don’t get upset.