
Table of Contents
- Stop sweeping the unknown under the carpet
- A glimpse into the gene pool without the carpet
- It is time for a change! Bring the unknown to the party!
- A conceptual framework to unify the known and the unknown in microbiome analyses
- Why gene clusters?
- Augmenting experimental data through a structured and contextualized coding sequence space
- What’s next?
Although it feels like yesterday, it has been three years since I gave feedback for the tongue-in-cheek blog post Microbial Dark Matter: The mullet of microbial ecology as someone who has been roaming in darker corners of our functional understanding of genes. I had agreed with this part of Meren’s post:
...the dark side is the best side as the many secrets of the microbial life hide in the dark corners of complex metagenomes for us to continue exploring...
The purpose of this blog post is to summarize the journey I did with Chiara Vanni, Matt Schechter, Tom Delmont, Meren, Martin Steinegger, Albert Barberán and Pier Luigi Buttigieg and others exploring the dark side, the science that emerged from it (check our preprint here), and my opinions on the future.
This story goes back in 2007-2008 when I was doing my Ph.D. at the Center of Advanced Studies of Blanes somewhere in Spain, close to the Mediterranean sea. Back then, together with my friend Albert Barberán we thought we would be able to develop methods to infer the function of the uncharacterized genes in our genomic and metagenomic analyses. How naive and wrong we were… At that time we were having fun playing with networks and graphical models. We had the “brilliant” idea of inferring co-occurrence networks using clusters of uncharacterized genes combined with Pfams and we were expecting to find associations between the known and unknown fraction and assign function following a guilty by association approach. Simultaneously, our colleague Pier Luigi Buttigieg at the MPI for Marine Microbiology in Bremen did something similar. Although it looked promising, we started to understand that this was not the way to go and that we were missing something essential, but we didn’t know what.
One day it dawned upon us, we were mixing apples and oranges! The whole field was designed and centered to work with what is known, and the unknown almost always was left on the side. Of course, we have attempts like DUFs in Pfam, or the category S in COG, but not a systematic integration of the unknown and the known, no common language to bridge the gap. Once we figured this out, it was only a matter of time until we had the necessary technology, enough data, and an awesome group of collaborators to start to develop a conceptual framework to unify the known and unknown coding sequence space.
First, the data have arrived. Large metagenomic projects such as GOS, HMP, and TARA, started to be public, and an avalanche of new genomes started to emerge in public databases owing to our ability to resolve genomes from metagenomes. Fortunately, advances in computational strategies had started to cope with large volumes of data. CD-HIT was our first choice to finally implement this idea. Yet with this clustering algorithm, the workflow we had prototyped was still taking months to cluster 300M of genes down to 30% of identity, even on a cluster of computers. Each clustering attempt took months leaving NO MARGIN OF ERROR. A condition under which developing and testing new workflows is nearly impossible.
One lucky day, somewhere around 2015-2016 during one of my GitHub fishing sprees (I like to search for random keywords in GitHub search) I stumbled across a code repository with a very promising tool. Although it was not so intuitive to use at that time, I gave it a try… and I couldn’t believe the results… this tool had just clustered the same 300M genes in ONE DAY! This tool was MMseqs2. Thanks to the help of Martin Steinegger and the MMseqs2 team we could make our plans a reality. Finally, we would be able to make mistakes and test different parameters to obtain the best representation of the unknown coding space.
In the same year, I got to know Lois Maigninen, Meren and Tom through our common friend Meghan Chafee. This wasn’t only the start of a scientific collaboration to work with the unknowns but a friendship. Since 2017, we have been meeting every autumn in Brest for the EBAME workshop, where we developed many of the ideas I will show you later. You can see a few of us enjoying Brittany.

It all began in my Ph.D., and now it ends with the brilliant work of my Ph.D. student Chiara Vanni and former MSc student Matt Schechter at the Max Planck Institute for Marine Microbiology. And of course, a nice group of friends and collaborators who also like the taste of slow-cooked science. If you are wondering what all of this is about, keep reading and learn more about our take on bringing light to the Dark side of genomes and metagenomes.
Brace yourself, this is not going to be a short post…
Stop sweeping the unknown under the carpet
What I am going to show is another attempt to provide a framework to make sense of a large number of genes of unknown function present in genomes and metagenomes, but with a twist. Before I continue, I would like to make one important point clear regarding our purpose:
We are not predicting function and we are not even trying. Predicting function from sequence similarity methods is not a trivial task and in many cases, you might be wrong. We are not referring to the assignment of a specific function like an oxidoreductase based on pieces of evidence like Pfam, but for example, the role of an enzyme in a metabolic context. You should always be suspicious of your metabolic reconstructions, and particularly when you are using short reads where you have partial matches. An example of how difficult it is to predict function even for well-characterized enzymes are the ones with high promiscuity. Also, the work of Frances H. Arnold in applying directed evolution to enzymes helps to understand how we are oversimplifying a very complex process (Arnold 1998; Arnold 2018).
With this in mind, let’s go back to business.
The unknown is a hot topic in microbiome studies. For many years, several groups have attempted to resolve the function of uncharacterized genes by combining biochemistry and crystallography (Jaroszewski et al., 2009); using environmental co-occurrence (Buttigieg et al., 2013); by grouping those genes into evolutionarily related families (Yooseph et al., 2007; Bateman et al., 2010; Sharpton et al., 2012; Brum et al., 2016; Wyman et al., 2018); and using remote homologies (Lobb et al., 2015; Bitard-Feildel & Callebaut, 2017). In 2018, Morgan N. Price and collaborators developed a high-throughput experimental pipeline that provides mutant phenotypes for thousands of bacterial genes of unknown function being one of the most promising methods to tackle the unknown. As you can see, many research groups have already been working on shedding light on genes with unknown functions. So, when we don’t even promise to provide you with functions for your mysterious genes, what are we doing then?
We are here to warn you…. WE ARE IN TROUBLE… and fortunately we’re all in this together… To help you stop having to sweep the unknown under the carpet, we are simply removing the carpet.
A glimpse into the gene pool without the carpet
Multiple lines of evidence suggest that soon we will be buried by sequences of genes of unknown function. The top graph below shows the number of genomes that have been deposited to the public space between 2000 to 2017, according to GTDB r86. We can see that metagenome-assembled genomes (MAGs) are a game-changer, and the number of genomes is starting to sky-rocket. And what is also sky-rocketing?
UNKNOWNS!
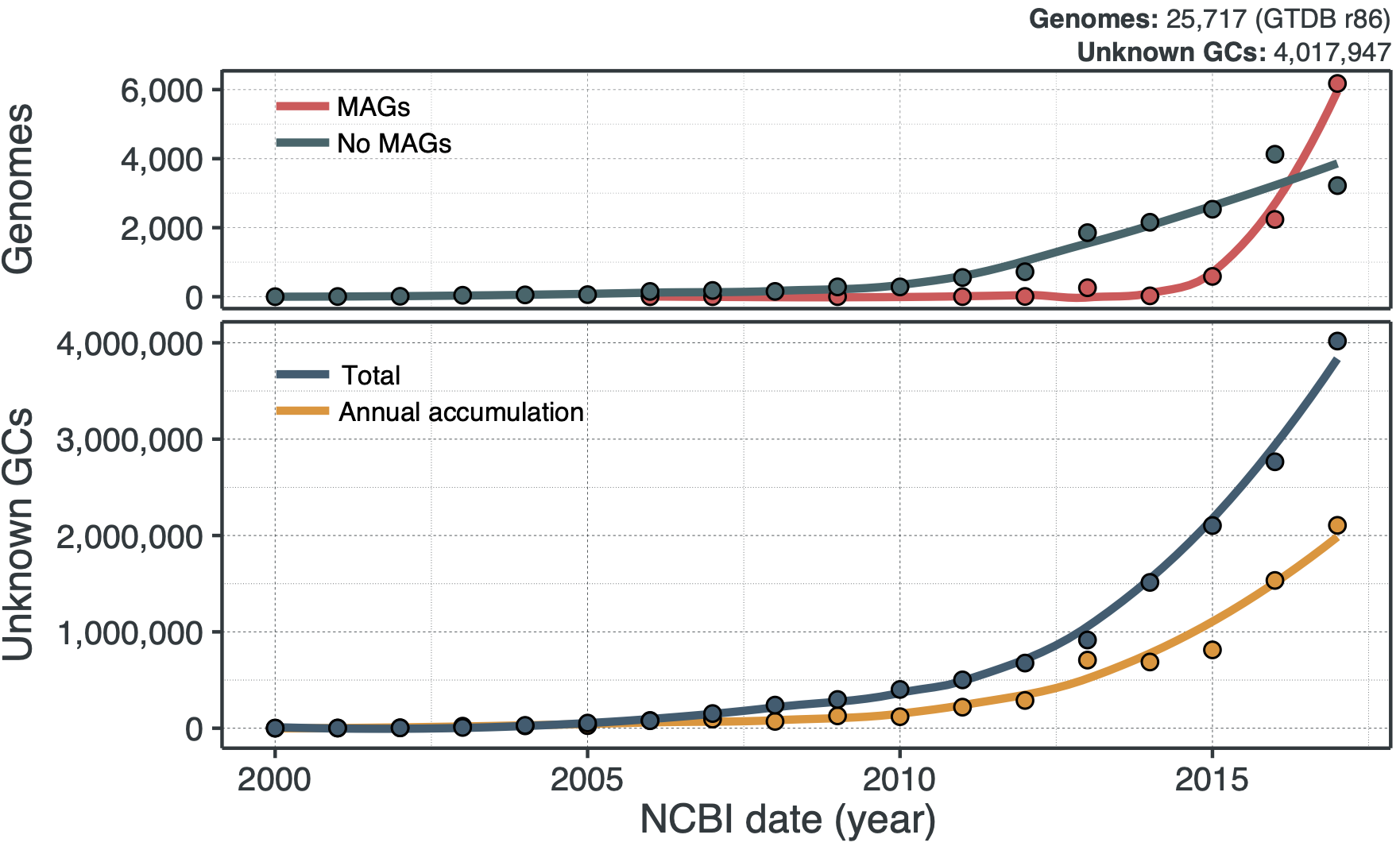
The bottom panel shows the number of new gene clusters (GCs) of known and unknown functions that have been accumulated since the early 2000s. At the moment, the rate of accumulation of the new GCs of unknown function is already 2X faster, and we are just at the beginning of the microbiome revolution. Wait a second, gene clusters? Weren’t we talking about genes here? I will come to this later, but for the time being, think of gene clusters as a unit to characterize the known and unknown in the microbial coding sequence space. I will be using gene clusters from now on as our approach is based on them. Trust me, all will make sense. Going back to the trends in the curves, MAGs seem to be one of the factors that influenced the accumulation of unknowns in recent years. When we look at the ratios of known and unknown in the different phyla present in GTDB we can see a clear pattern, where the phyla with more unknowns are the ones with the larger number of MAGs as shown in the following plot:
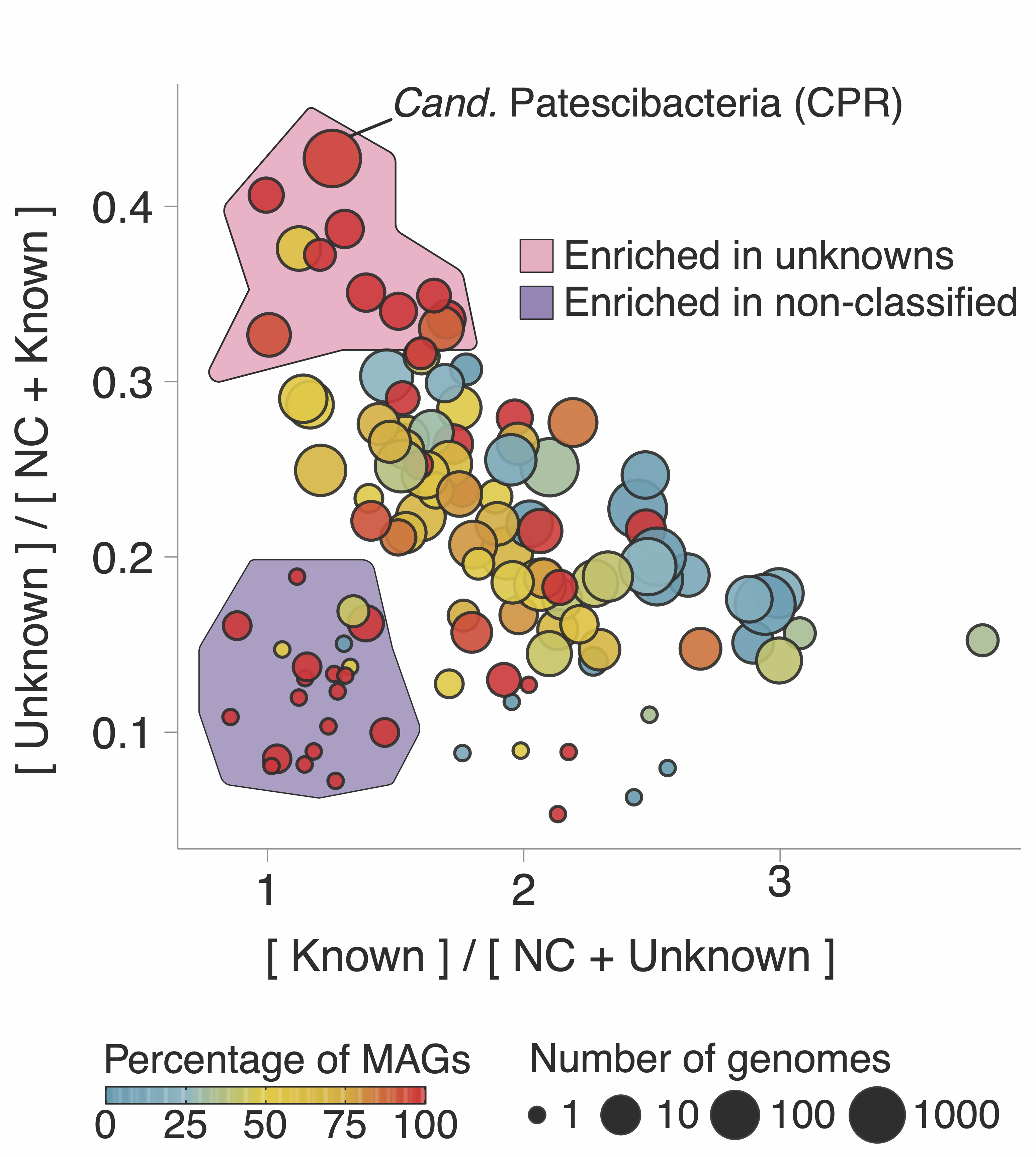
There is also an interesting area in the plot, the phyla enriched with something called “Enriched in non-classified” (NC). I will not enter into the details here, but our approach provides methods to identify very novel genomes, and of special interest to the microbiome community, our methods can be used to identify contaminants in MAGs. I will write about this in another post, promise. Let’s go back to the plot. There is one phylum that stands out, the Cand. Patescibacteria or more commonly known as Candidate Phyla Radiation (CPR), a phylum that has raised considerable interest due to their unusual biology.
Here, we provide a collection of 54,350 GCs of unknown function at different taxonomic level resolutions which will be a valuable resource for the advancement of knowledge in the CPR research efforts.
So, this is just the beginning… What will happen in ten years when new sequencing technologies give us access to all those uncharted regions of microbial diversity we are just dreaming today? It is time to act now before it is too late. We cannot keep sweeping the unknown under the carpet, especially when the devil is in the detail.
Most of the unknown is lineage-specific and at the species level. GTDB provides the perfect phylogenomic playground to test hypotheses, and we used a similar approach than the one used in Annotree. We identified those gene clusters that are lineage-specific, ruling out the ones that might have a prophage origin, and map them on the GTDB phylogenomic tree.
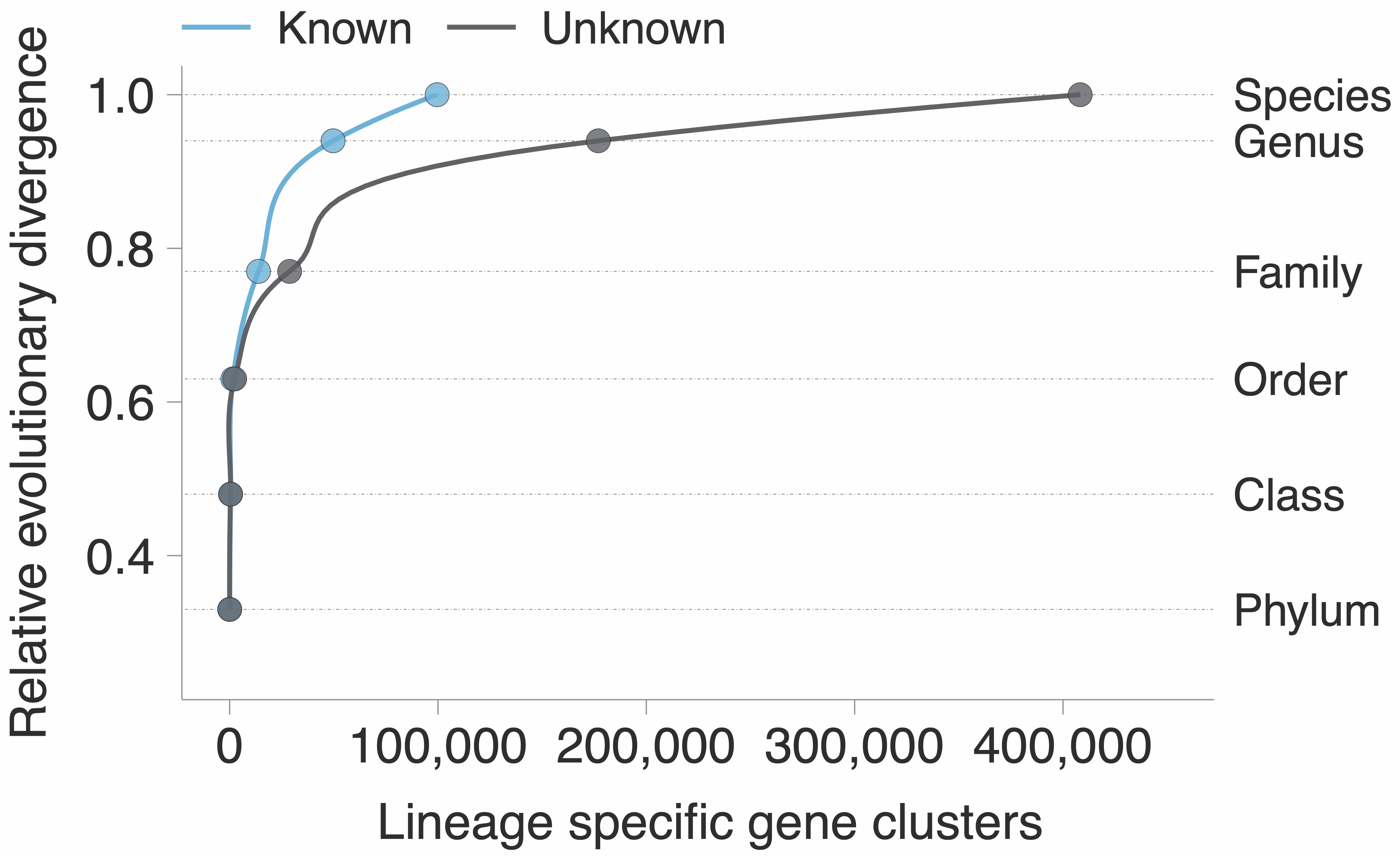
The figure speaks for itself, there is such a vast amount of unknown biology that defines what a microbe is, and most of the time we are not doing anything useful with it. Or even worse, we just discard it. When we look at the environment, we have a similar picture to what we have shown already in a genomic context. At the moment we explored the extent of the unknown in marine and human microbiomes as shown in the plot below.
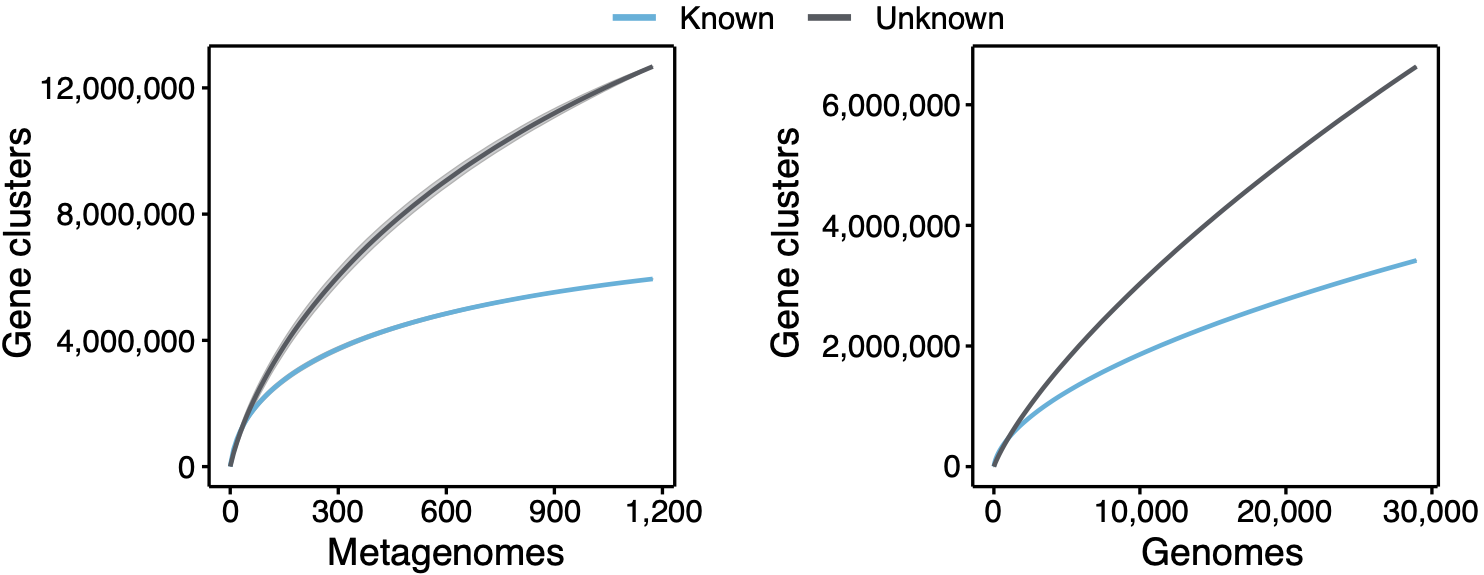
Even though we explored two of the best-characterized microbiomes, the curve of the unknown is far from reaching a plateau. We are looking forward to having our hands in the soil and other microbiomes. Looking at the genomic collector curves one can have an idea of what is waiting for us. Exciting times! The following plot shows the proportion of gene clusters that have a broad or a narrow distribution in marine and human metagenomes. The majority of the unknown gene clusters have a narrow distribution being most of the time sample-specific.
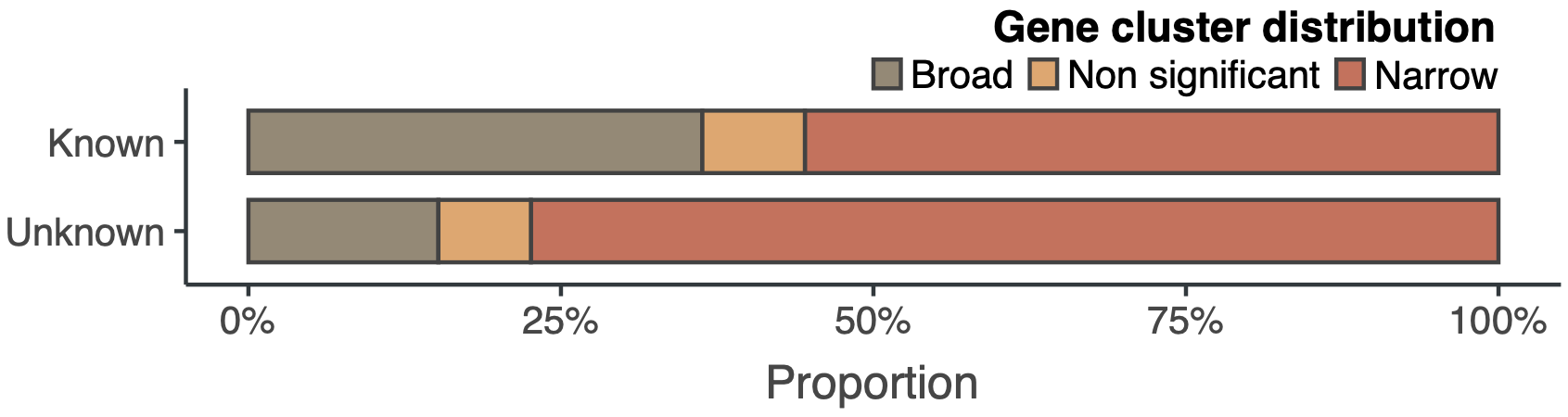
Here is where we can start to hypothesize about the importance of the unknown in the processes of niche adaptation owing to its lineage-specificity and sample-specificity. But unfortunately, we still don’t know the underlying mechanisms, or how all those unknowns emerge, but at least, now we have somewhere to start. Thus far we have been showing a few insights about the unknown fraction in genomes and metagenomes. By now, you might wonder, where are all these results coming from? What gene clusters have to do with it? At least, we hope that we convinced you about the importance of the unknowns and that now you are eager to know how we manage to integrate the unknown fraction in microbiome analyses. Let’s dive into it!
It is time for a change! Bring the unknown to the party!
Current analytical approaches for genomic and metagenomic data generally do not include the uncharacterized fraction. This is a problem as their results are constrained to conserved pathways and housekeeping functions. This inability to handle shades of the unknown is an immense impediment to realizing the potential for discovery of microbial genomics and microbiology at large. So let’s get to the root of these limitations. Most of you are familiar with the prototypical genomic and metagenomic analysis workflow, just in case, it can be briefly summarized in the following steps.
While this workflow has been extremely useful for many years, it is time for an upgrade! Here it goes:
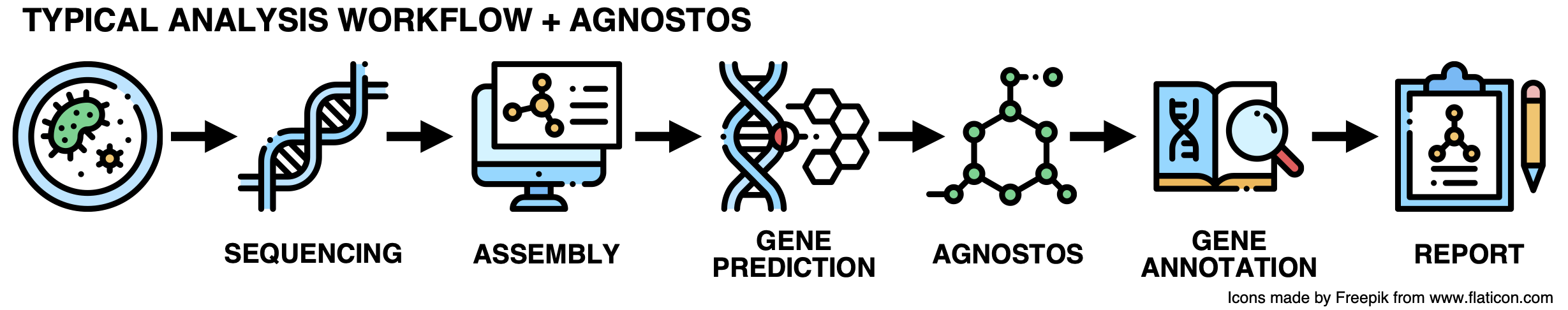
All problems solved! Let’s move on to the next challenge. Jokes aside, what is Agnostos?
AGNŌSTOS THEOS. The phrase agnōstōn theōn (nominative singular, agnōstos theos) was found inscribed on Greek altars dedicated "to the unknown gods." The inscription had no mystical or theosophical meaning, but arose out of a concern for cultic safety: no one wanted to incur the wrath of gods whose names were unknown but who just might exist and be vexed by the lack of honors
As we don’t want to incur the wrath of SCIENCE leaving the unknown aside, we developed a conceptual framework and a computational workflow to unify the known and unknown coding sequence space (CDS-space) in microbiome analyses. AGNOSTOS integrates into the traditional workflow transparently, providing the opportunity of exploiting the full set of genes in your genome and metagenome. It synthesizes the conceptual and technical foundations we created to unify the known and unknown and provide a practical solution to one of the most significant ongoing challenges in microbiome analyses.
A conceptual framework to unify the known and the unknown in microbiome analyses
We have been spending some time to figure out a way on how to include the unknown sequence space in our analyses and simultaneously cover genomes and metagenomes. It all starts by finding the proper concepts that can help in partitioning the CDS-space. We started many years ago using a derivative from the famous phrase from Donald Rumsfeld:
(...) Reports that say that something hasn't happened are always interesting to me, because as we know, there are known knowns; there are things we know we know. We also know there are known unknowns; that is to say we know there are some things we do not know. But there are also unknown unknowns—the ones we don't know we don't know. And if one looks throughout the history of our country and other free countries, it is the latter category that tend to be the difficult ones (...)
Back then, we split the genes in our dataset in the KNOWN, the KNOWN UNKNOWN and the UNKNOWN UNKNOWN. While this might sound cool, it is not intuitive at all and doesn’t capture the real nature of the data we were trying to describe. As I already said before, we were young and naive. But in 2014, after presenting our current work at ISME, Jed Fuhrman suggested changing those terms for the ones we are using now. So we passed from tongue twisters to something that is really descriptive and helped us to develop our ideas in a more coherent manner. Now we partition the CDS-space in four categories, two for the known and two for the unknown:
- Known with Pfam annotations: genes annotated to contain one or more Pfam entries (domain, family, repeats or motifs) but excluding the domains of unknown function (DUF)
- Known without Pfam annotations: which contains the genes that have a known function but lack a Pfam annotation. Here we can find intrinsically disordered proteins or small proteins among others.
- Genomic unknown: genes that have an unknown function (DUF are included here) and found in sequenced or draft genomes
- Environmental unknown: genes of unknown function not detected in sequenced or draft genomes, but only in environmental metagenomes or metagenome-assembled genomes.
Our whole approach develops around those four main concepts. With them, we are able to bridge genomics and metagenomics and simultaneously unify the known and unknown CDS-space based on its level of darkness. We introduce a subtle change in the traditional workflow and this is where AGNOSTOS shines. Our objective is to provide the best representation of the unknown space and we gear all our efforts towards finding sequences without any evidence of known homologies by pushing the search space beyond the twilight zone of sequence similarity. We will travel deeper into the twilight zone in the future using Hydrophobic Cluster Analysis, this is just the beginning. We combine this exhaustive search for homology with the partitioning of the CDS-space using gene clusters as the fundamental units. Then we enrich each of the gene clusters with lots of different types of information. The following figure summarizes how the conceptual model and the computational workflow complements each other.
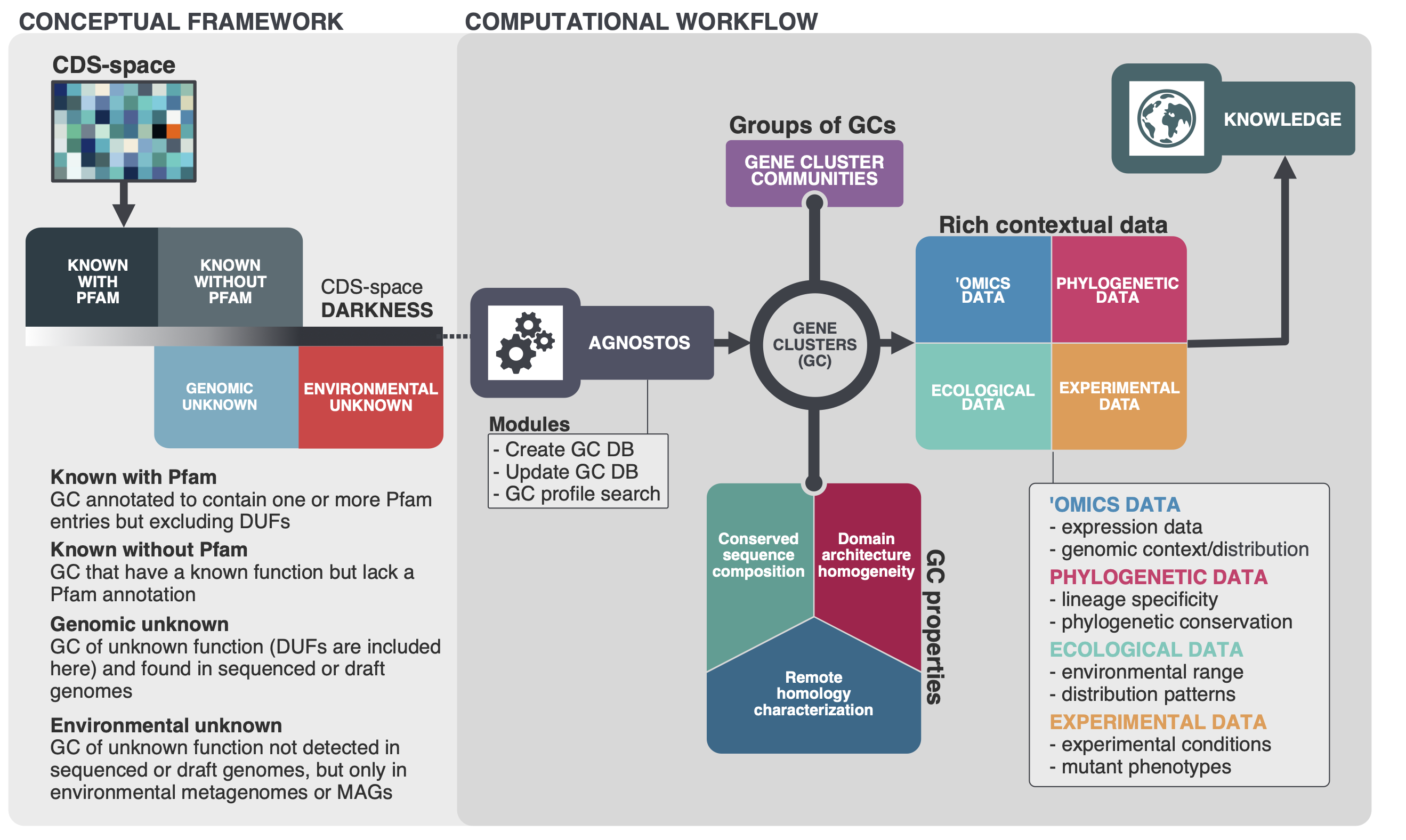
One of our top priorities when dealing with the unknown is adding as much context as we can. Information is power. We bring together different types of information providing a powerful tool to generate hypotheses with the ultimate goal to augment experimental data. Later I will show you an example of all the different parts working together. But first, you might be still wondering why we are using gene clusters.
Why gene clusters?
The reason why we are using gene clusters is purely practical. At the moment, there aren’t any other well-tested methodologies to partition the CDS-space. We have been referring several times about partitioning the CDS-space, but what does this mean? The figure below summarizes our vision and how the gene clusters are used to connect the conceptual framework with the conceptual workflow.
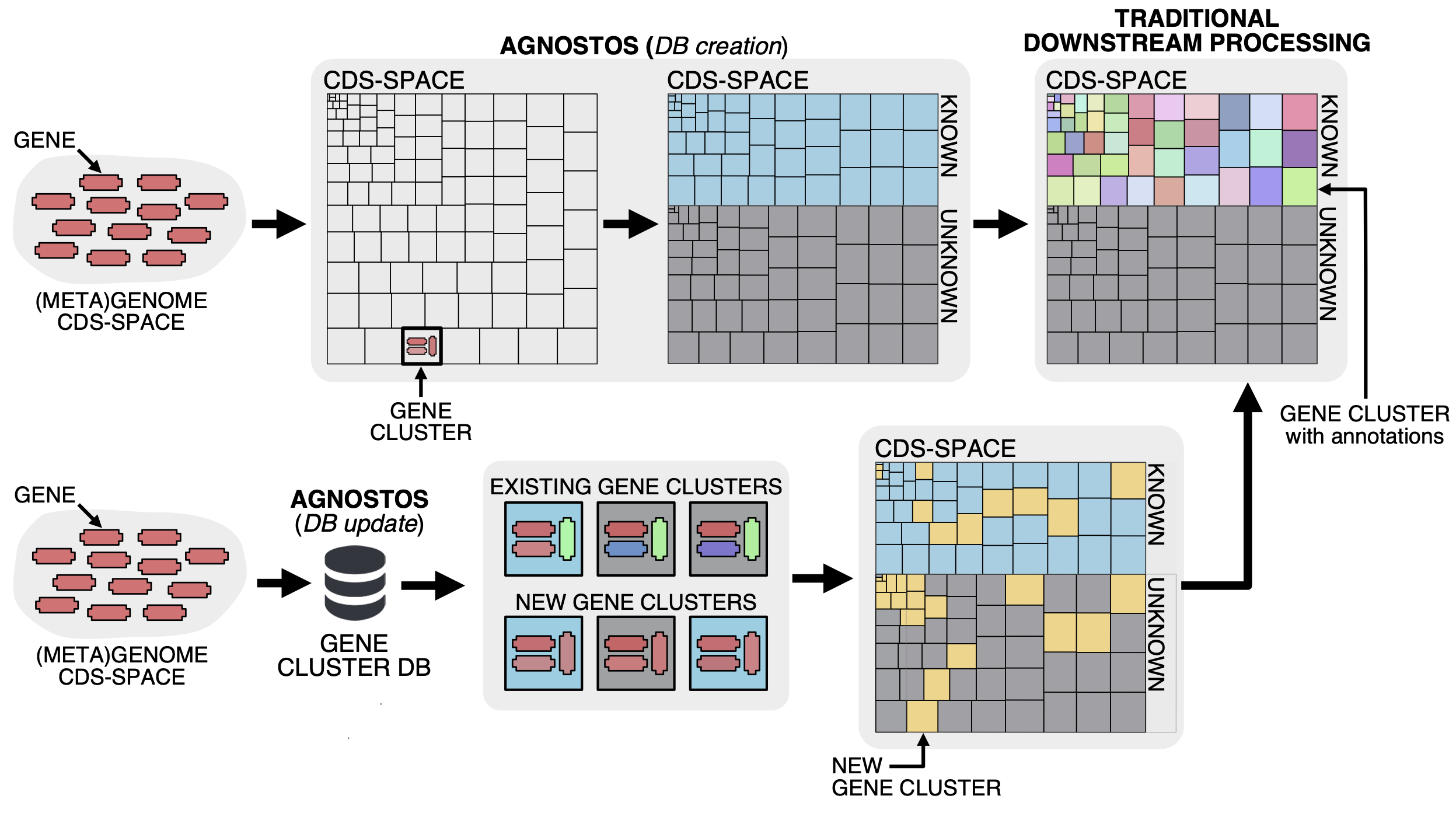
Our conceptual framework relies on defining a basic unit that can bring together genomes, environment, and at the same time group similar genes together. We use AGNOSTOS to achieve these objectives and structure the CDS-space using gene clusters in a mechanistic manner minimizing the number of assumptions. When the CDS-space is already structured, we identify the known and unknown regions. Afterward, we can add any other layer of information and analyze the known fraction with the traditional workflows. With this procedure we can integrate the unknown to the known as both fractions share the same unit, the gene cluster. Furthermore, AGNOSTOS uses the ability of MMseqs2 to update existing gene clusters providing a scalable continuous integration of new data.
Gene clusters, besides being useful to reduce and structure a dataset, can be a powerful tool when used wisely. With the clustering we bring together genes that share at least an identity of the 30%, just before the limits of the twilight zone and where sequence similarity starts to get messy. We think clustering at this identity is a good threshold, as we are in a mid-point where we can perform analyses at different scales and also bring together genomes and the environment. Think of it as a microscope where you can zoom in and zoom out, and where we use the contextual data associated with each gene for staining.
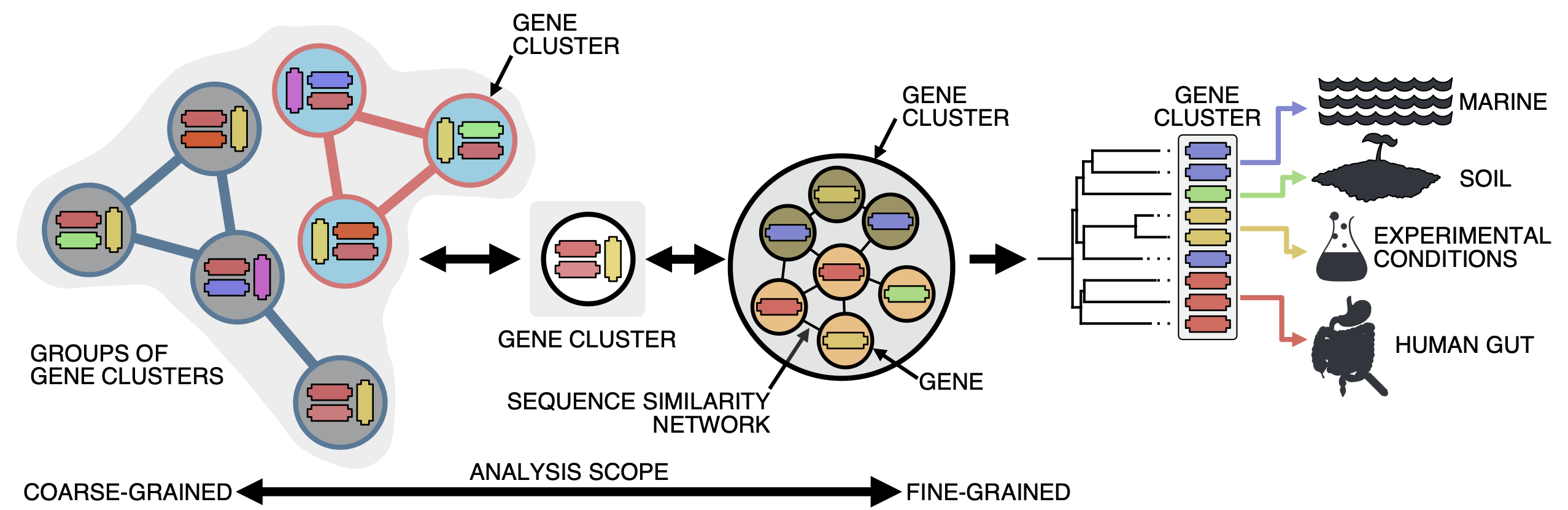
When we zoom-in, we can exploit the gene associations within each gene cluster for fine-grained analyses; and when we zoom out, we get groups of gene clusters (we call them gene cluster communities) based on their shared homologies for coarse-grained analyses. And as we can track the origin of each gene in a gene cluster, we can obtain a contextual landscape that spans across genomes and environments. To get to this point, first we need to obtain extremely high-quality gene clusters and AGNOSTOS takes care of it. We have developed different methods to assure that the gene clusters we use have a highly conserved intra-homogeneous structure, both in terms of sequence similarity and domain architecture homogeneity. You can read more about it here.
In a not so distant future most probably we will not use gene clusters. With the new developments in deep learning we will be able to use the large corpora of proteins to define a language model embedding for protein sequences. Then, we might be able to use those models to build a narrative of the CDS-space and provide a more compelling partitioning. Here is where the simplicity and the beauty of our conceptual framework lays, it is independent on how we partition the coding space.
But at the end of the day, the most important fact is… is this useful for something? Or is it just a collection of fuzzy words and speculations?
Augmenting experimental data through a structured and contextualized coding sequence space
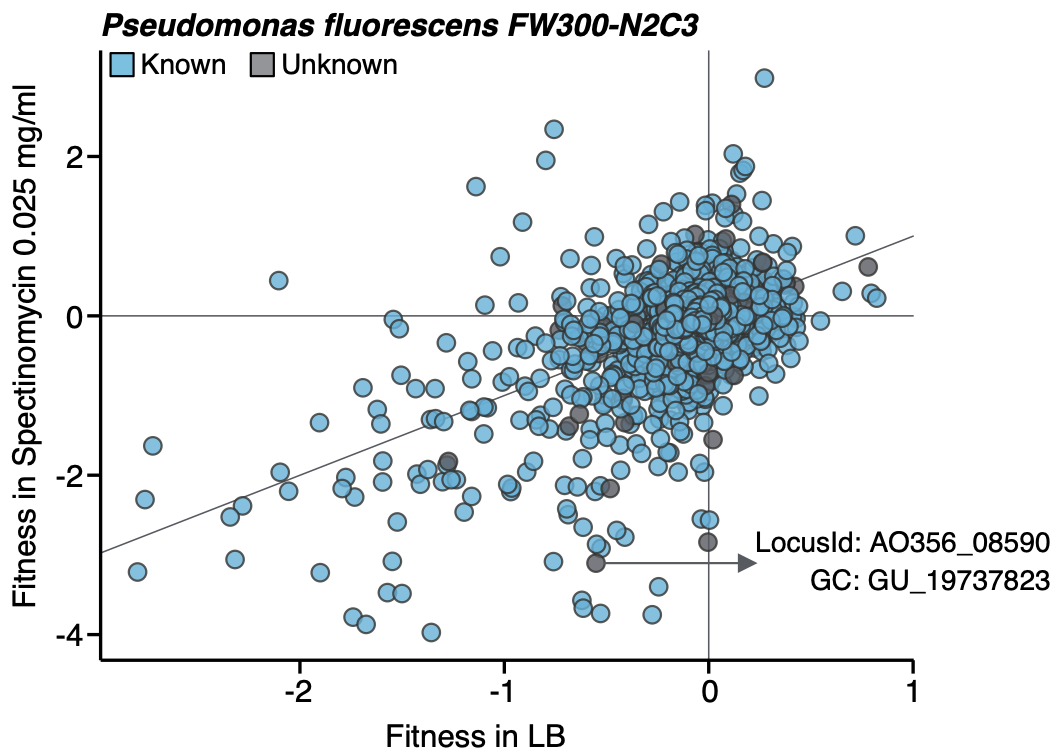
While our approach has been useful to provide one of the clearest pictures of the status of the unknown in microbial systems, its real power reveals when you combine it with experimental data. We selected one of the experimental conditions tested in Price et al. to demonstrate the potential of our approach to augment experimental data. We compared the fitness values in plain rich medium with added Spectinomycin dihydrochloride pentahydrate to the fitness in plain rich medium (LB) in Pseudomonas fluorescens FW300-N2C3. This antibiotic inhibits protein synthesis and elongation by binding to the bacterial 30S ribosomal subunit and interferes with the peptidyl tRNA translocation. As shown in the figure below, we identified a gene with unknown function with locus id AO356_08590 to present a strong phenotype (fitness = -3.1; t = -9.1) in this experimental condition. This gene belongs to our genomic unknown space and it is a member of the gene cluster GU_19737823.
Now we can use the microscope that we previously described to zoom out and explore the gene clusters associated with GU_19737823. This gene cluster is a member of the GU_c_21103 community:
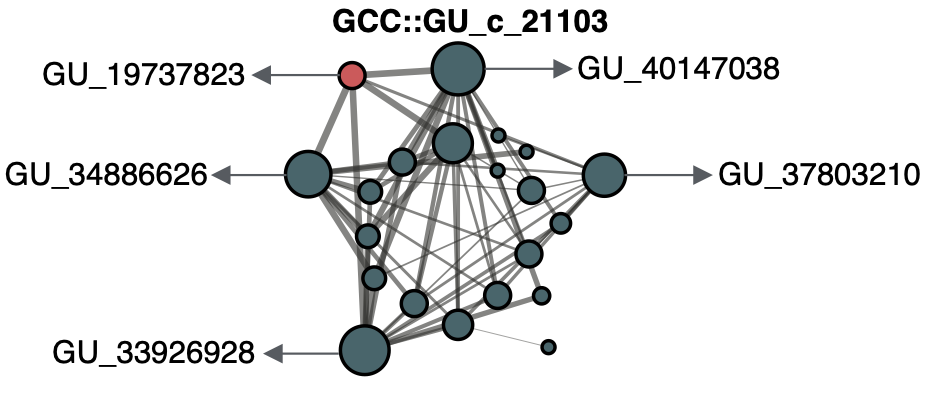
If you are interested in how we create the groups of gene clusters, or how we call them gene cluster communities, have a look here.
This gene cluster community has twenty-one gene clusters that can have a genomic or/and environmental origin. In this case, five of them can be mapped to a genome in GTDB r86 and easily we can retrieve all homologous genes in these gene clusters and explore their genomic neighborhoods.
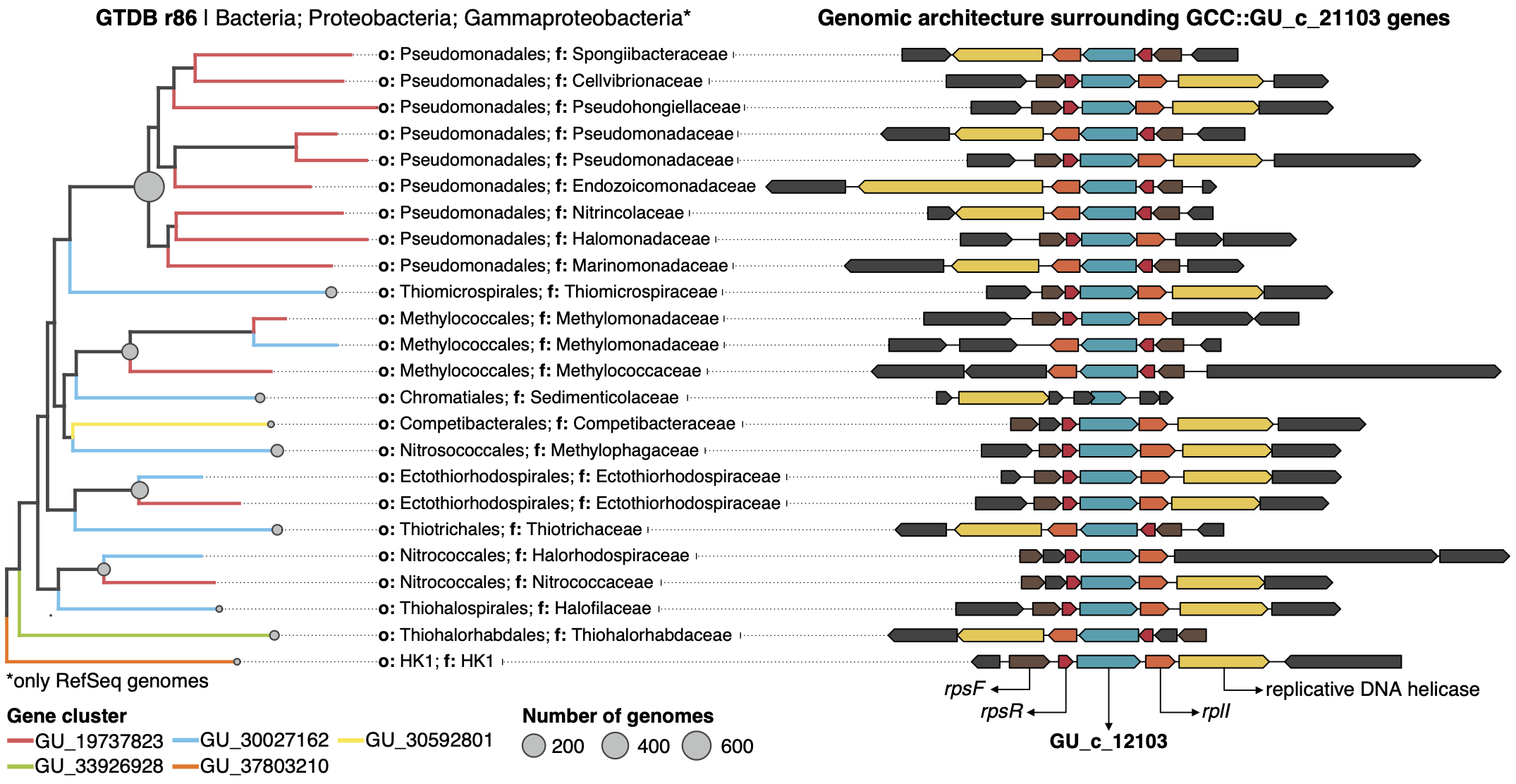
The first thing we notice is that all the members of GU_c_21103 are constrained to the class Gammaproteobacteria, and interestingly GU_19737823 is mostly exclusive to the order Pseudomonadales. The gene order in the different genomes analyzed is highly conserved, finding GU_19737823 after the rpsF::rpsR operon and before rpll. rpsF and rpsR encode for 30S ribosomal proteins, the prime target of spectinomycin. The combination of the experimental evidence and the associated data inferred by our approach provides strong support to generate the hypothesis that the gene AO356_08590 might be involved in the resistance to spectinomycin. And of course, these findings are not only limited to this genome, but to all the others that have a homologous in the gene cluster community GU_c_21103. We can go one step further and close the circle by looking at the distribution of GU_19737823 in the environment.
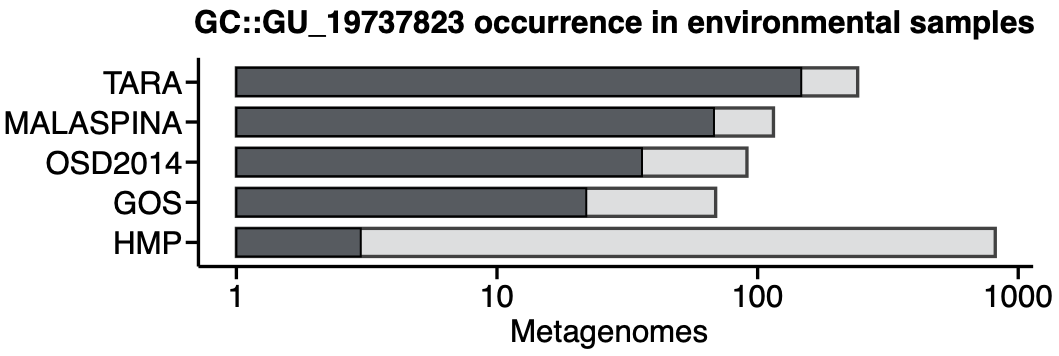
Darker bars show how many of the metagenomes (clear bars) contain genes members of the gene cluster GU_19737823. As expected, this gene cluster is mostly found in non-human metagenomes as Pseudomonas are common inhabitants of soil and water environments. This is the reason why finding this gene cluster in human related samples can be very interesting, owing to the potential association of P. fluorescens and human disease where Crohn’s disease patients develop serum antibodies to this microbe.
Pretty cool, isn’t it?
What’s next?
The long road to have the framework and workflow in place is paying out now, we have a powerful tool to explore and learn from the unknown. We started to explore the unknown fraction of the soil environment together with Albert Barberán, Hannah Holland-Moritz and Noah Fierer. We are developing new methods to identify novel biosynthetic gene clusters using the genomic unknowns in collaboration with Marnix Medema. And maybe the most unexpected outcome of our approach is the ability to identify contaminants in MAGs. Together with Tom Delmont, Meren Martin Steinegger and Chiara Vanni we are exploring new ways to help with the manual curation of large collections of MAGs (and reference databases) by identifying problematic contigs.
However, we still have to face many challenges. Some are technical, like the limited recovery of genes from the environment owing to the limitations of assembly and gene prediction. This is why alternatives like protein-level assembly combined with the exploration of the assembly graphs’ neighborhoods become very attractive for our purposes. While others are related to the scientific community. As of today, there is no community consensus on what it means to be an unknown. If you ask around, you will quickly realize that most of us who have thought about this question will have a different concept of it.
One of my highest priorities is to incentivize the scientific community to build a common effort to define the different levels of the unknown and address the actual problematic that our science is only based, in the best of the cases, on using only 50% of the available data. We can do better! We should work together on defining clear guidelines and protocols, provide controlled vocabularies and maybe even create a GSC standard or a MIxS extension. What is clear, is that our work proves that the integration of the unknown fraction is possible and I expect that it can help to bring a brighter future for microbiome sciences.
We look forward to hearing your opinions and suggestions, drop a comment or send an email to dark@metagenomics.eu
Learn more reading our preprint or explore our website.