Pangenomics, Phylogenomics, and ANI of Spiroplasma genomes
Table of Contents
Summary
The purpose of this document is to provide a reproducible workflow for the pangenomics, phylogenomics, and ANI of 31 Spiroplasma genomes. It also includes the detailed description of how we linked all these analyses together into the following figure using anvi’o:
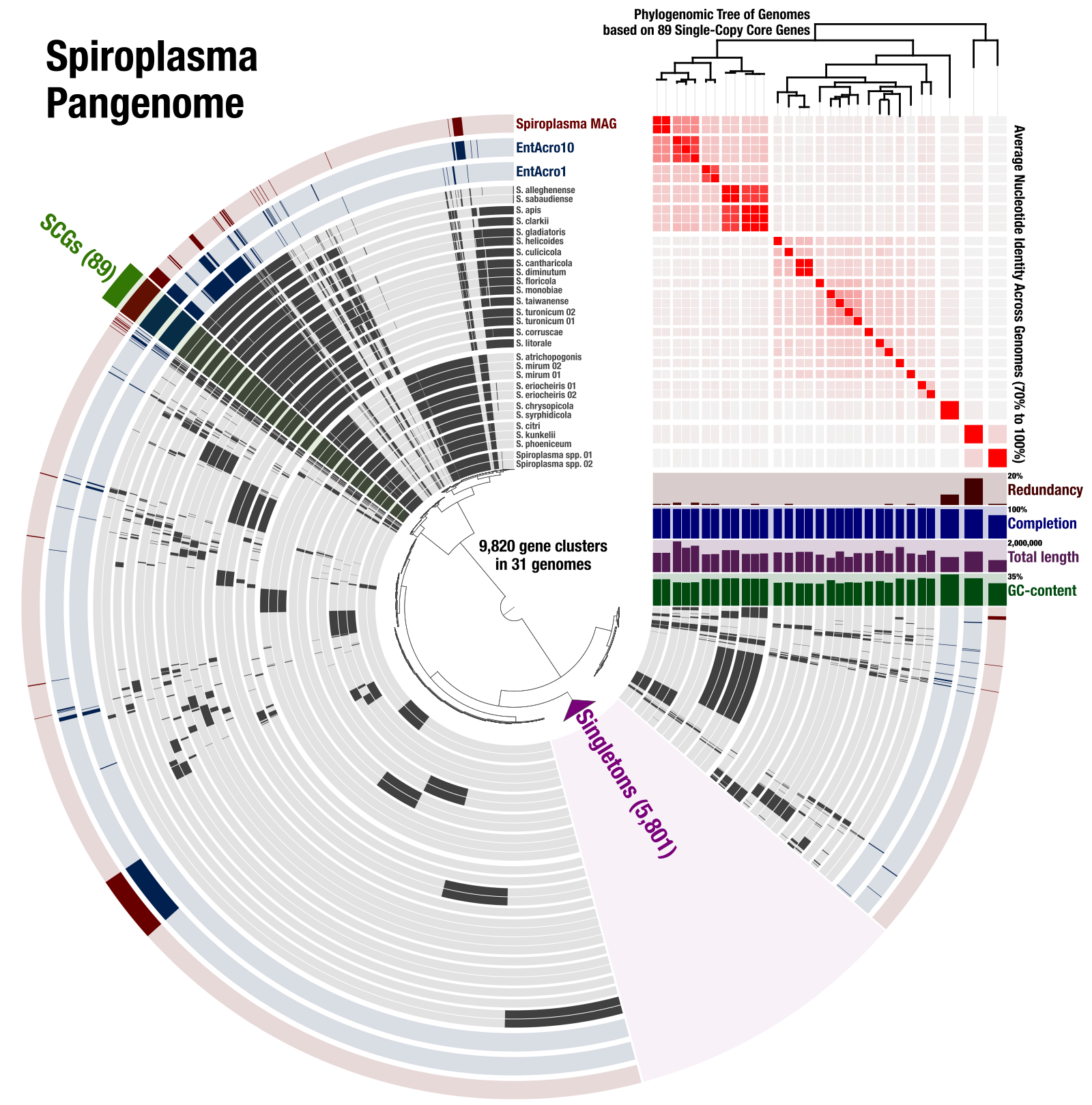
doi:10.6084/m9.figshare.8201852 gives access to anvi’o files to visualize, update, and edit the Spiroplasma pangenome.
If you have any questions, please feel free to leave a comment down below, send an e-mail to us, or get in touch with other anvians through Discord:

Introduction
This is a part of the collaboration we had with Carl Yeoman and his colleagues.
Our team here at the Meren Lab processed the raw metagenomic sequences Yeoman et al generated from Wheat Stem Sawfly, reconstructed and manually curated a novel population genome that resolved to the Spiroplasma genus, and put this genome into the context of other Spiroplasma genomes that are available in public databases.
The Spiroplasma genome had a GC% content of 24.56%, 754 open reading frames, components of a single ribosomal RNA operon, and 23 tRNA encoding genes. Full annotation and the genome are available through PATRIC and the NCBI Bioproject PRJNA540284.
To put this genome into the context of other Spiroplasma genomes we used genomes available through the NCBI and included recently published genomes from the Ixodetes clade of mollicutes by Sapountzis et al (named EntAcro1 and EntAcro10).
We tested this workflow on anvi’o v5.5 and it should work with anvi’o releases v5 or later. To see the installation instructions of anvi’o please visit here.
Metagenome assembled genomes
If you wish to fully reproduce the workflow down below, you can download the genomes we have used for this study the following way:
# create a directory for MAGs
mkdir MAGs && cd MAGs
# download Yeoman et al. Spiroplasma genome
curl -L -O http://merenlab.org/data/spiroplasma-pangenome/files/Spiroplasma_MAG.fa.gz
# download Sapountzis et al. genomes
curl -L -O http://merenlab.org/data/spiroplasma-pangenome/files/Entomoplasmatales_EntAcro1.fa.gz
curl -L -O http://merenlab.org/data/spiroplasma-pangenome/files/Entomoplasmatales_EntAcro10.fa.gz
# unpack all and go to the upper directory
gzip -d * && cd ..
Pangenomics, Phylogenomics, and ANI
To determine the relationship of the Spiroplasma MAG to previously published Spiroplasma genomes we first downloaded all genomes from the NCBI that resolve to genus Spiroplasma (following the protocol described in the blog post “Accessing and including NCBI genomes in ‘omics analyses in anvi’o”:
ncbi-genome-download --assembly-level complete \
bacteria \
--genus Spiroplasma \
--metadata metadata.txt
Then we processed the resulting GenBank files to prepare them for an anvi’o analysis:
anvi-script-process-genbank-metadata -m metadata.txt \
-o Spiroplasma \
--output-fasta-txt Spiroplasma-fasta.txt \
--exclude-gene-calls-from-fasta-txt
We then removed contigs that are shorter than 1,000 nucleotides from the two genomes EntAcro1 and EntAcro10,
anvi-script-reformat-fasta ./MAGs/Entomoplasmatales_EntAcro1.fa \
-l 1000 \
-o ./MAGs/Entomoplasmatales_EntAcro1-min1K.fa
anvi-script-reformat-fasta ./MAGs/Entomoplasmatales_EntAcro10.fa \
-l 1000 \
-o ./MAGs/Entomoplasmatales_EntAcro10-min1K.fa
And created a fasta.txt (the format of which is described here) to run the anvi’o pangenomic workflow:
echo -e "Spiroplasma_MAG.fa\t`pwd`/MAGs/Spiroplasma_MAG.fa" \
>> Spiroplasma-fasta.txt
echo -e "Entomoplasmatales_EntAcro10\t`pwd`/MAGs/Entomoplasmatales_EntAcro10-min1K.fa" \
>> Spiroplasma-fasta.txt
echo -e "Entomoplasmatales_EntAcro1\t`pwd`/MAGs/Entomoplasmatales_EntAcro1-min1K.fa" \
>> Spiroplasma-fasta.txt
After editing this file to make sure names look human-readable (a copy of the final input file is here), we generated a configuration that looked like this (a copy of it is here),
{
"fasta_txt": "Spiroplasma-fasta.txt",
"project_name": "Spiroplasma",
"external_genomes": "external-genomes.txt"
}
Note that the external-genomes.txt file will be generated automatically by the pangenomic workflow.
And finally ran the anvi’o pangenomic workflow (with 6 cores) to get the pangenome computed:
anvi-run-workflow -w pangenomics \
-c pan-config.json \
--additional-params \
--jobs 6 \
--resources nodes=6
This took about 15 minutes on a laptop computer. This workflow generated a new directory in our working space called 03_PAN, which included the pan database and genomes storage files.
To add a heatmap that shows the average nucleotide identity estimates across genomes, we run the following command, which uses PyANI program in the background and adds the ANI information into the pan database automatically:
anvi-compute-ani -e external-genomes.txt \
-o ANI \
-p 03_PAN/Spiroplasma-PAN.db \
-T 6
If you are using anvio v6 or later, anvi-compute-ani has been replaced by anvi-compute-genome-similarity. The above command remains the same otherwise.
To infer evolutionary associations between 31 genomes in our pangenome, we used single-copy core genes (SCGs) across all genomes for a phylogenomic analysis. To recover a FASTA file for individually aligned and concatenated SCGs specific to the pangenome, we ran the following command:
anvi-get-sequences-for-gene-clusters -p 03_PAN/Spiroplasma-PAN.db \
-g 03_PAN/Spiroplasma-GENOMES.db \
--min-num-genomes-gene-cluster-occurs 31 \
--max-num-genes-from-each-genome 1 \
--concatenate-gene-clusters \
--output-file 03_PAN/Spiroplasma-SCGs.fa
This resulted in a FASTA file, which we first cleaned up by removing nucleotide positions that were gap characters in more than 50% of the seqeunces using trimAl,
trimal -in 03_PAN/Spiroplasma-SCGs.fa \
-out 03_PAN/Spiroplasma-SCGs-clean.fa \
-gt 0.50
And ran the phylogenomic analysis using IQ-TREE with the ‘WAG’ general matrix model to infer a maximum likelihood tree:
iqtree -s 03_PAN/Spiroplasma-SCGs-clean.fa \
-nt 8 \
-m WAG \
-bb 1000
In order to organize genomes in the pangenome during the visualization step, we generated a ‘layers order’ file (the format of which is explained here), and imported it into the pan database:
# generate the file
echo -e "item_name\tdata_type\tdata_value" \
> 03_PAN/Spiroplasma-phylogenomic-layer-order.txt
# add the newick tree as an order
echo -e "SCGs_Bayesian_Tree\tnewick\t`cat 03_PAN/Spiroplasma-SCGs-clean.fa.contree`" \
>> 03_PAN/Spiroplasma-phylogenomic-layer-order.txt
# import the layers order file
anvi-import-misc-data -p 03_PAN/Spiroplasma-PAN.db \
-t layer_orders 03_PAN/Spiroplasma-phylogenomic-layer-order.txt
At this point all the information was in the database, so we visualized it using the following command.
anvi-display-pan -p 03_PAN/Spiroplasma-PAN.db \
-g 03_PAN/Spiroplasma-GENOMES.db
Which opened an interactive interface with the pangenome.
Polishing the pangenome
We find it critical to properly visualize complex data, and often put the extra effort to prepare and polish our visualizations for publication.
This figure shows the initial/raw display of the pangenome:
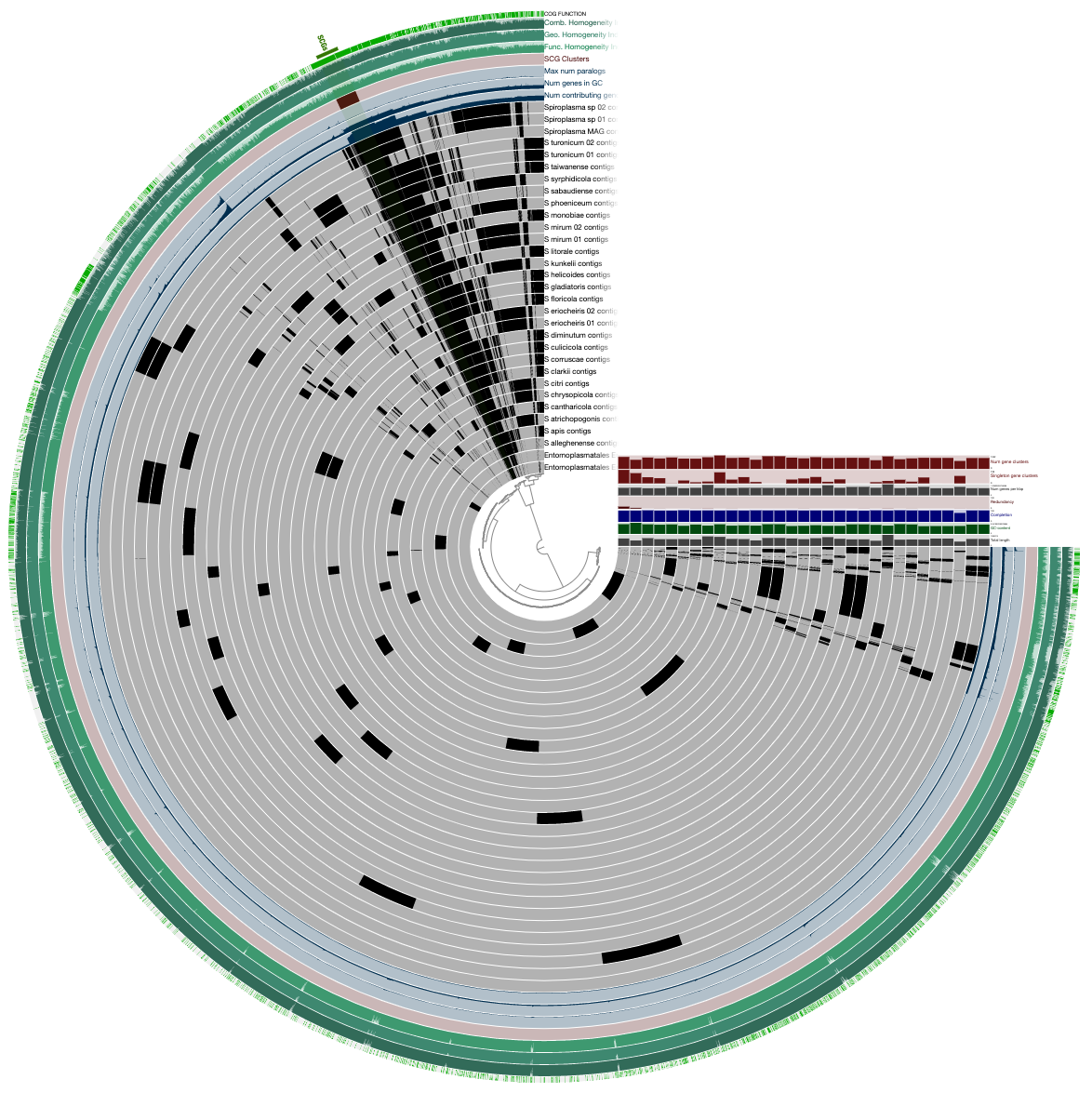
We polished this display by displaying (1) the phylogenomic tree and (2) the ANI estimates we computed and stored in the pan database prevoiusly through the following steps:
- Click Draw after the interface first opens.
- Click Settings > Layers > Layer Groups > ANI_percentage_identity to display the ANI heatmap.
- Select all ANI layers by selecting ANI_percentage_identity from Settings > Layers > Select all layers in a group.
- Increase the minimum values for each ANI layer all at once by entering 0.7 in Settings > Layers > Edit attributes for multiple layers > Min.
- Click Settings > Layers > Redraw layer data to see changes.
- Select ‘SCG_Phylogeny’ from Settings > Layers > Order by combo box to order genomes by the phylogenomic tree
- Increase the radius of the dendrogram in the center by entering 6000 in Settings > Main > Show Additional Settings > Dendrogram > Radius
- Increase the height of the phylogenomic tree on the right-top by entering 2000 in Settings > Layers > Tree/Dendrogram > Height
- Increase the size and selections layer by entering 400 in Settings > Main > Additional Settings > Selections > Height
- Click Settings > Main > Show Additional Settings > Custom margins to enable custom margins
- Use Settings > Main > Layers to increase genome / group distances using the margin column
- Command-right-click to the branch of singletons and click Collapse from the menu
- Reduce the opacity of layer bacgrounds by setting 0.15 to Settings > Main > Show Additional Settings > Layers > Background opacity
- Increase the maximum font size by entering 240 in Settings > Main > Show Additional Settings > Layer Labels > Max. Font Size
- Click Draw to see changes
Which gave us this display:
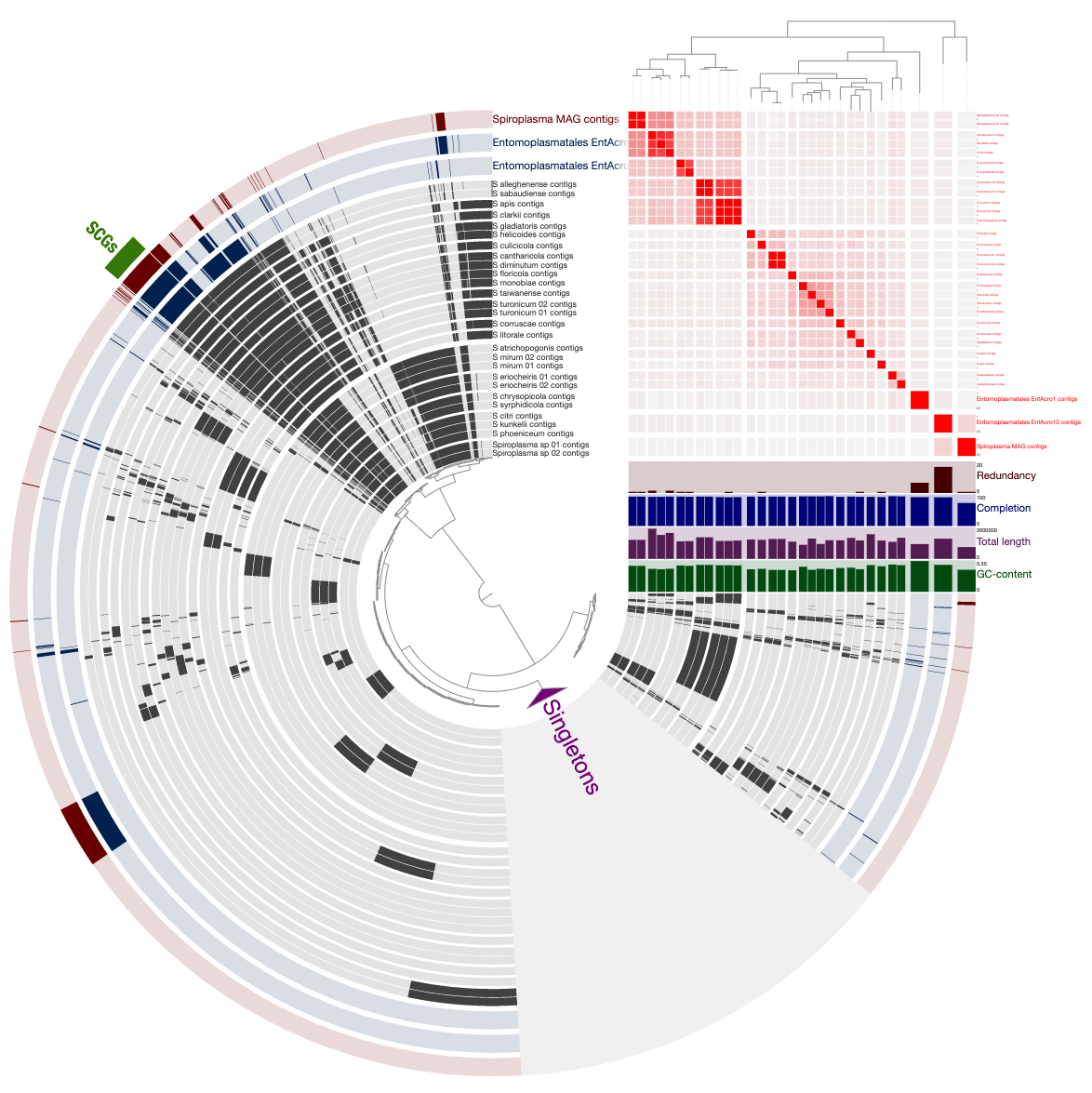
We then used the Save SVG button that is located in the Settings panel (bottom-right) to download the SVG file, and further polished labels in Inkscape to acquire this final figure:
doi:10.6084/m9.figshare.8201852 gives access to anvi’o files to visualize, update, and edit the Spiroplasma pangenome.
If you have any questions, please feel free to leave a comment down below, send an e-mail to us, or get in touch with other anvians through Discord: