A tutorial on pangenome graphs in anvi'o


Table of Contents
The purpose of this reproducible tutorial is to describe step-by-step how gene-centric pangenome graphs are computed, interactively visualized, and summarized for downstream analyses in anvi’o.
Are you only here to see the pangenome graph?
If you are here only to visualize the products of this tutorial on your computer, please install anvio-dev on your computer following the instructions here, and run the following commands on your terminal:
# go to your home directory
cd
# create a new directory and go into it
mkdir UNDATIPELAGIBACTER-PAN-GRAPH && cd UNDATIPELAGIBACTER-PAN-GRAPH
# download all necessary files
curl -L https://cloud.uol.de/public.php/dav/files/TN2bxBCbAS5DRDJ -o UNDATIPELAGIBACTER-GENOMES.db
curl -L https://cloud.uol.de/public.php/dav/files/8eZZYqNrAdXF4TA -o UNDATIPELAGIBACTER-PAN-GRAPH.db
# initiate the interactive interface
anvi-display-pan-graph
For those of you who are here to go through the tutorial, please continue :)
Throughout this tutorial we will use 29 genomes that belong to Undatipelagibacter (a.k.a. SAR11 subclade Ia.3.VI), a recently named genus in the family Pelagibacteraceae.
The tutorial will start with raw FASTA files for each genome, and end with a pangenome graph and associated flat-text summaries of genomic regions identified in them for downstream analyses. We will demonstrate how to
- Re-orient FASTA files using anvi-reorient-genomes,
- Turn reoriented FASTA files into contigs-db files using anvi-gen-contigs-database,
- Annotate the resulting contigs-dbs with useful information using anvi-run-hmms, anvi-run-ncbi-cogs, anvi-run-kegg-kofams, and anvi-run-pfams,
- Compute a pangenome from these contigs-dbs using anvi-pan-genome,
- Compute a pangenome graph using the pan-db using anvi-pan-genome-graph,
- Visualize the resulting pan-graph-db using anvi-display-pan-graph, and,
- Summarize the pangenome graph into flat-text files for downstream analyses using anvi-summarize.
An interactive (but read-only) access to the final pangenome graph is available at https://merenlab.org/upg-interactive/
Why pangenome graphs?
If you are familiar with the anvi’o pangenomics workflow, you already know how to identify which genes are shared across a set of genomes and which are unique. But conventional pangenome analyses collapse genes into unordered clusters, discarding their chromosomal context. A pangenome graph preserves the syntenic relationships between genes: it tells you not only which genes are shared, but where they sit relative to each other on the chromosome. This makes it possible to precisely delineate structurally conserved backbone regions from variable regions, and to study how genomic neighborhoods differ across organisms. In short, a conventional pangenome answers “what genes do these genomes have?”, while a pangenome graph answers “how are these genomes organized?”.
This tutorial serves two purposes. The first one is to familiarize you to how pangenome graphs are implemented in anvi’o and walk you through each step for you to be able to apply it to your own datsaets. The second one is to generate the input files for a reproducible bioinformatics workflow for Henoch et al (2026) paper that officially introduces the gene-centric pangenome graph toolkit in anvi’o and its application to Undatipelagibacter. If you are interested in our downstream analyses of pangenome graphs and the kinds of insights they offer, please consider also reading our paper “Synteny-aware microbial pangenomes reveal blueprints of genomic variation”.
You can follow this tutorial with your own FASTA files, or using the FASTA files that are included below. If you have any questions, you can send an email, or find @alexh4180 and/or @meren.anvio at

Setting up the work environment
You will need two things to go through this tutorial:
- A working anvi’o installation
- FASTA files for your genomes
The next two subsections will help you ensure both of these needs are satisfied appropriately before you begin with the rest of the tutorial.
Anvi’o setup
The software tools for pangenome graphs in anvi’o are quite new and are not yet available with any of the major anvi’o releases. However, you can follow the instructions at https://anvio.org/install/ to install the development version of anvi’o (a.k.a., anvio-dev) on your computer.
If you are seeing a similar output when you run the following command on your terminal, you are golden:
anvi-self-test -v
Anvi'o .......................................: eunice (v9-dev)
Python .......................................: 3.10.15
Profile database .............................: 42
Contigs database .............................: 24
Pan database .................................: 21
Pangraph database ............................: 4
Genome data storage ..........................: 8
Structure database ...........................: 2
Metabolic modules database ...................: 4
tRNA-seq database ............................: 2
Genes database ...............................: 6
Auxiliary data storage .......................: 2
Workflow configurations ......................: 5
You also need to make sure that you have all the necessary annotation databases accessible on your system since some of the steps below will require them. If you have just installed anvi’o, or if you had it before but never ran any of the following programs, please run them in your terminal for anvi’o to set things up for you to avoid issues later:
anvi-setup-ncbi-cogs
anvi-setup-pfams
anvi-setup-kegg-data
If you are not sure, just run them anyway since if everything is already setup, anvi’o will tell you something along the lines of “things already seem to be set up – use the --reset flag if you want to download and rebuild everything from scratch”.
Data setup
Whether you will be using your own FASTA files, or those that this tutorial will provide, it is a good idea to start fresh in a work directory. Let’s first create a work directory for this tutorial:
mkdir ~/pangraph-tutorial/
cd ~/pangraph-tutorial/
We will do everything in this -currently empty- directory.
Download the FASTA files:
curl -L https://cloud.uol.de/public.php/dav/files/jSFTXG3cSQMBjYX -o FASTA-FILES-ORIGINAL.tar.gz
Unpack the archive and get rid of the tar.gz file:
tar -xzf FASTA-FILES-ORIGINAL.tar.gz && rm FASTA-FILES-ORIGINAL.tar.gz
Now we have all the FASTA files in the newly unpacked FASTA-FILES-ORIGINAL directory, and the rest of the tutorial will make use of this structure. Just to make sure we are on the same page at this point, this is how our current work directory looks like:
.
└── FASTA-FILES-ORIGINAL
├── HIMB122.fa
├── HIMB140.fa
├── HIMB1488.fa
├── HIMB1491.fa
├── HIMB1493.fa
├── HIMB1506.fa
├── HIMB1507.fa
├── HIMB1513.fa
├── HIMB1518.fa
├── HIMB1526.fa
├── HIMB1552.fa
├── HIMB1556.fa
├── HIMB1559.fa
├── HIMB1573.fa
├── HIMB1577.fa
├── HIMB1593.fa
├── HIMB1597.fa
├── HIMB1611.fa
├── HIMB1631.fa
├── HIMB1636.fa
├── HIMB1641.fa
├── HIMB1662.fa
├── HIMB1685.fa
├── HIMB1701.fa
├── HIMB1702.fa
├── HIMB1723.fa
├── HIMB1758.fa
├── HIMB1765.fa
└── HIMB1770.fa
2 directories, 29 files
If you are intending to follow this tutorial with your own FASTA files and if you would like the rest of the tutorial to work for you seamlessly, please consider putting your FASTA files in a FASTA-FILES-ORIGINAL directory, and keep in mind the following critical suggestions:
- Give your FASTA files a one-word names without any spaces or funny characters (you can safely use
_character, and everything in this tutorial should still work). - Make sure your FASTA files have the extension
.fain lowercase, and not.fasta,.fna, or any other extension. - We assume that your FASTA files for this analysis correspond to relatively closely related organisms. Pangenomes can be computed for various levels of taxonomy, and one can go as far as an entire phylum for some branches of life. But pangenome graphs are much more conservative. We advise you to remain below genus level for your initial analyses. It is difficult to give precise numbers that will work equally well throughout the tree of life, but a minimum gANI above 90% would be a good starting expectation. In the case of Undatipelagibacter the average ANI was around 95%, and it went as low as 93%.
- This tutorial assumes that all your FASTA files represent complete genomes with a single chromosome. Working with pangenome graphs with fragmented genomes is also possible, but since preserved and explicit gene synteny is one of the critical features of this workflow, at this stage we are reluctant to encourage such applications. That said, please feel free to get in touch with us so we can share our best practices and two cents with you for a useful analysis of your genomes.
If you are not sure whether every single FASTA file you have has a single contig, you can always run this command and expect to see only number 1 next to each file:
for i in FASTA-FILES-ORIGINAL/*fa; do echo "$i: $(grep -c '>' $i)"; done
If you are here, and if each of the statements above make sense, we are ready to start the actual analysis.
Reorienting genomes
This purpose of this step is to make sure all sequences start from an evolutionarily meaningful position. Because bacterial chromosomes are circular, the position where a sequencing or assembly project “breaks” the circle into a linear FASTA sequence is arbitrary. If two genomes start at completely different positions on the chromosome, the graph algorithm could misinterpret genuinely conserved regions as rearrangements (even though we have means to avoid that, it is better to be safe than sorry). By reorienting all genomes so they start at the same landmark such as the origin of replication or any other highly conserved region across all genomes), we ensure that the linear representations of these circular chromosomes are directly comparable before gene calling takes place.
Depending on how genomes are finalized, the starting point of each sequence may differ from one another. While graph building step can take care of rotations of genomes, it is best practice to align genomes first, which we can do with the anvi’o program anvi-reorient-genomes. It is imperative that this step is implemented before the genes are called in FASTA files, which typically takes place when FASTA files are turned into anvi’o contigs-db files.
We first need to create a fasta-txt file, which is a two-column TAB-delimited file that can be passed to anvi-reorient-genomes to let the program know where to find all FASTA files of interest. You can generate this file for our original FASTA files by running the following command in our work directory:
anvi-script-gen-fasta-txt --input-dir FASTA-FILES-ORIGINAL/ \
-o FASTA-FILES-ORIGINAL/fasta.txt
The resulting fasta.txt will look like this:
name |
path |
|---|---|
| HIMB122 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB122.fa |
| HIMB140 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB140.fa |
| HIMB1488 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1488.fa |
| HIMB1491 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1491.fa |
| HIMB1493 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1493.fa |
| HIMB1506 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1506.fa |
| HIMB1507 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1507.fa |
| HIMB1513 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1513.fa |
| HIMB1518 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1518.fa |
| HIMB1526 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1526.fa |
| HIMB1552 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1552.fa |
| HIMB1556 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1556.fa |
| HIMB1559 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1559.fa |
| HIMB1573 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1573.fa |
| HIMB1577 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1577.fa |
| HIMB1593 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1593.fa |
| HIMB1597 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1597.fa |
| HIMB1611 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1611.fa |
| HIMB1631 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1631.fa |
| HIMB1636 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1636.fa |
| HIMB1641 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1641.fa |
| HIMB1662 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1662.fa |
| HIMB1685 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1685.fa |
| HIMB1701 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1701.fa |
| HIMB1702 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1702.fa |
| HIMB1723 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1723.fa |
| HIMB1758 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1758.fa |
| HIMB1765 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1765.fa |
| HIMB1770 | /Users/meren/pangraph-tutorial/FASTA-FILES-ORIGINAL/HIMB1770.fa |
Now we can run the program to reorient all these genomes (which should take about 15 to 20 seconds on a laptop computer):
anvi-reorient-genomes --fasta-txt FASTA-FILES-ORIGINAL/fasta.txt \
--use-dnaa-for-reference-orientation \
--threads 8 \
-o FASTA-FILES-FINAL
This alignment step (which produces a ton of visualizations for you to enjoy) will store all FASTA files under a new directory, FASTA-FILES-FINAL. Here is a fun experiment to test if things worked as expected: running the following command to see the first 50 nucleotides of each genome in the directory FASTA-FILES-ORIGINAL,
for f in FASTA-FILES-ORIGINAL/*.fa; do awk '!/^>/ {print substr($0, 1, 50); exit}' "$f"; done
results in the following output (where each line is the first 50 nucleotides of one genome):
AAAAATGTCATTTTGGATGACAAACAAAATAAAAATAAAATTTTCTCAAA
TATTTTGGGTGACTATACGATTACCCTGAGAGATTTTTGTGTTAAATTAT
AGTTGATGTAAGCGCTTTAAAACCTGGTAACACAATTACAGTTTTATGGA
AAGAATTAGTTTTTACCGAAGGAAACTCAAGAGCAATAGATGATCAAAAA
GTTTTTGTATTTAAAATATATCCCTCTTGTTGAATTATTTTATCTAAGTC
AAAATAAATTGATTTTTAATTATTTTTAGTTTTGTAAAAAAATTTTGATT
TAACAAATAAAGCAACTTATTTTAAAAATGATGAAATCATTTTTACAGAA
AAAACATTGAAATATTTGCTGAAGAAATAATTTATGAAAAAAATAAAGAA
CTTAGTCATGGGGGGATTTTAAATAATTGTGAAGGTTCTATTTCACTTTT
ATTTACCACCACCTGCCGTTTCAAGTAAGGCACCAAATAAAACGAACAGA
AGCTTGTTTAAAGCTTTCTTCAGATTCTTTTAATTTATTTTGTTCATGCA
AATCTGAAGAAAGCTTTAAACAAGCTATATCATTAAAGCCAAACTATGCT
TTATTATAAATAAATTTATCAGCCTCTATAATAATTCCTTCATTATTCTT
AAATGGAAATGTAGTAATAGATGATAAAGTTAAAGGATTAAAATTATTTG
AATTAGTATACAAATAAATACTATTAATTACTTCTTTTTAAAATGTATTC
ATTTAATAAAATTAAATATTCAATATTGCTAATTTTATTTACTTTTTATT
AAATAAATGAATCAAGAGGTAACAATTCTTATATTAACTCATAAAAGTAA
ATATTAGGATAATAAAAAAAAGAGTAAATTAAATAATTATTTTTATTTTT
CTCATTTCTTTTAAATTCAAATTCTTTTGCAGTTATTATTTTTTCATCAA
GCAAAGTATTTAAATTTAAAATTAAAATGGAAGGGTACAGGAATCAATAC
TTAAAGCCAAACTATGCTGAAGCTTACAGTAACTTGGGTAATATACTACG
CCAAACTATGCTGAAGCTTACAGTAACTTGGGTAATACATTACAAGAACA
TTAAAGGATATAAATATTCTTTAAATAAAGATATATTTCAACTTATTCCA
TATTTGTGACCCTGGTGTGACACATATTTTCTATTTGAAGTGGTAAGGGG
TTAATTCACTATATTAAACTATTAAAATTTTCTTCGTTTTAGTGATAAAA
TTAGAAAACCGATGCTCTATCCAGCTGAGCTAAGGGCGCAATATTTGAAA
GAAATAATTGATAAAAAAACAAACAATCAATTATTCACAAACAAGATAAC
GTAAGTAGCTTGCTCTGAAAAAATTTTATATTTATTTATCTGATCTTCTA
TTATTGAAGTTCAAAAAAATTCTTACAAAGAATTAACTGAATTCTATGCT
In contrast, running this command to see the first 50 nucleotides of each genome in the directory FASTA-FILES-FINAL,
for f in FASTA-FILES-FINAL/*.fa; do awk '!/^>/ {print substr($0, 1, 50); exit}' "$f"; done
results in the following output:
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTAAAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGTTTCAGAGGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGACGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAACAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGTTTCAGAGGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
ATGAATAAATCTTTTACAAATAAAAATATTAATAATTTTGCTTCAGAAGA
Demonstrating that anvi-reorient-genomes did its job: the near-identical starting sequences across all 29 genomes correspond to the DnaA region near the origin of replication, confirming that all chromosomes are now anchored at the same evolutionarily conserved position. We are ready to move on.
Generating contigs-db files
Download the contigs-db files instead?
If you want to skip this section, you can simply download the contigs-db files generated in this chapter by running the steps listed below. For that, please run the following lines in your terminal:
curl -L https://cloud.uol.de/public.php/dav/files/7bRpYznDNBedSRk -o CONTIGS-DBs.tar.gz
tar -xzf CONTIGS-DBs.tar.gz && rm CONTIGS-DBs.tar.gz
If you are here, it means you have your final FASTA files in the FASTA-FILES-FINAL directory. Next, is to make sure we have an anvi’o contigs-db file for each of the FASTA files.
Generating contigs-db is quite straightforward. For a single FASTA file, there are three things one typically needs to do: (1) fix FASTA deflines, (2) generate a contigs-db, and (3) annotate it with functions. To apply all these steps to all FASTA files in the FASTA-FILES-FINAL directory, here we can use a for loop in our terminal, since loops make everything much more fun and dynamic.
While this tutorial explains the following steps by running each tool one after another, our group typically generates contigs-db files and annotates them using the program anvi-run-workflow with the ‘contigs’ workflow. The anvi-run-workflow uses snakemake in the background, and it allows us to run a large number of tasks in parallel on our HPC environment, making life much easier. But this strategy makes the FASTA to contigs-db journey like a black box, so here in this tutorial we go through each step one by one to make sure what is really happening behind the scenes is more obvious.
Here is the for loop that will turn each FASTA file under FASTA-FILES-FINAL into a contigs-db and store it under a new CONTIGS-DBs directory:
If you are following this tutorial word-by-word, you must run the script below from within ~/pangraph-tutorial/.
# first, let's get rid of any CONTIGS-DBs directory we may have in our work
# directory that may be left behind from a previous attempt
rm -rf CONTIGS-DBs
# create a fresh CONTIGS-DBs directory so we store everything in it
mkdir CONTIGS-DBs
# let's create a variable that keeps all our FASTA files we are
# interested in, so we can 'loop through' them next:
fasta_files=( FASTA-FILES-FINAL/*.fa )
# now we are ready to go through each FASTA file, turn them into
# anvi'o contigs-db files, and annotate them with functions
# and HMMs. This is the part that will take some time and it is
# best to run it on an HPC
num_threads="8"
for fasta_file in "${fasta_files[@]}"
do
# learn the genome name without the file extension
genome=$(basename $fasta_file .fa)
# simplify the headers. this script will simply ensure
# that the FASTA deflines are anvi'o compatible. since
# the original deflines are in the matching FASTA files
# under FASTA-FILES-ORIGINAL, we are not losing any
# information here
anvi-script-reformat-fasta $fasta_file \
--simplify-names \
--prefix $genome \
--overwrite-input
# this anvi'o program will generate a contigs database
# for this file. note that it is reading from one directory
# and writing its output into another
anvi-gen-contigs-database -f FASTA-FILES-FINAL/$genome.fa \
-o CONTIGS-DBs/$genome.db
# the following programs will annotate the newly
# generated contigs-db file with (1) the default HMMs
# ship with anvi'o, (2) COGs from NCBI, (3) KOfams from
# KEGG, (4) Pfams from .. well Pfams, and will also call
# rRNA and tRNA genes:
anvi-run-hmms -c CONTIGS-DBs/$genome.db -T $num_threads
anvi-run-ncbi-cogs -c CONTIGS-DBs/$genome.db -T $num_threads
anvi-run-kegg-kofams -c CONTIGS-DBs/$genome.db -T $num_threads
anvi-run-pfams -c CONTIGS-DBs/$genome.db -T $num_threads
anvi-scan-trnas -c CONTIGS-DBs/$genome.db -T $num_threads
done
If you are here, regardless of whether you downloaded your contigs-db files, or generated them from scratch, you should see something like this when you look into the CONTIGS-DBs directory:
CONTIGS-DBs/
├── HIMB122.db
├── HIMB140.db
├── HIMB1488.db
├── HIMB1491.db
├── HIMB1493.db
├── HIMB1506.db
├── HIMB1507.db
├── HIMB1513.db
├── HIMB1518.db
├── HIMB1526.db
├── HIMB1552.db
├── HIMB1556.db
├── HIMB1559.db
├── HIMB1573.db
├── HIMB1577.db
├── HIMB1593.db
├── HIMB1597.db
├── HIMB1611.db
├── HIMB1631.db
├── HIMB1636.db
├── HIMB1641.db
├── HIMB1662.db
├── HIMB1685.db
├── HIMB1701.db
├── HIMB1702.db
├── HIMB1723.db
├── HIMB1758.db
├── HIMB1765.db
└── HIMB1770.db
1 directory, 29 files
And you should se an output like this if you were to run anvi-db-info on any of the contigs-db files in that directory:
anvi-db-info CONTIGS-DBs/HIMB122.db
DB Info (no touch)
===============================================
Database Path ................................: HIMB122.db
description ..................................: [Not found, but it's OK]
db_type ......................................: contigs (variant: unknown)
version ......................................: 24
DB Info (no touch also)
===============================================
project_name .................................: HIMB122
contigs_db_hash ..............................: hash09d8a1e8
split_length .................................: 20000
kmer_size ....................................: 4
num_contigs ..................................: 1
total_length .................................: 1453515
num_splits ...................................: 72
gene_level_taxonomy_source ...................: None
genes_are_called .............................: 1
external_gene_calls ..........................: 0
external_gene_amino_acid_seqs ................: 0
skip_predict_frame ...........................: 0
splits_consider_gene_calls ...................: 1
scg_taxonomy_was_run .........................: 0
scg_taxonomy_database_version ................: None
trna_taxonomy_was_run ........................: 0
trna_taxonomy_database_version ...............: None
reaction_network_ko_annotations_hash .........: None
reaction_network_kegg_database_release .......: None
reaction_network_modelseed_database_sha ......: None
reaction_network_consensus_threshold .........: None
reaction_network_discard_ties ................: None
creation_date ................................: 1778571941.70956
modules_db_hash ..............................: 68221bd12b30
gene_function_sources ........................: KEGG_Module,KEGG_Class,COG24_FUNCTION,KEGG_BRITE,COG24_CATEGORY,Pfam,COG24_PATHWAY,KOfam,Transfer_RNAs
* Please remember that it is never a good idea to change these values. But in some
cases it may be absolutely necessary to update something here, and a
programmer may ask you to run this program and do it. But even then, you
should be extremely careful.
AVAILABLE GENE CALLERS
===============================================
* 'pyrodigal-gv' (1,509 gene calls)
* 'Transfer_RNAs' (30 gene calls)
* 'Ribosomal_RNA_16S' (1 gene calls)
AVAILABLE FUNCTIONAL ANNOTATION SOURCES
===============================================
* COG24_CATEGORY (1,278 annotations)
* COG24_FUNCTION (1,278 annotations)
* COG24_PATHWAY (513 annotations)
* KEGG_BRITE (1,042 annotations)
* KEGG_Class (329 annotations)
* KEGG_Module (329 annotations)
* KOfam (1,043 annotations)
* Pfam (2,775 annotations)
* Transfer_RNAs (30 annotations)
AVAILABLE HMM SOURCES
===============================================
* 'Archaea_76' (76 models with 32 hits)
* 'Bacteria_71' (71 models with 70 hits)
* 'Protista_83' (83 models with 3 hits)
* 'Ribosomal_RNA_16S' (3 models with 1 hit)
* 'Ribosomal_RNA_18S' (1 model with 0 hits)
* 'Transfer_RNAs' (68 models with 30 hits)
If all looks good on your end, we can move on to the next step.
Computing the pangenome
Download the Undatipelagibacter pangenome instead?
If you want to skip this section, you can simply download the final products of the analysis steps listed below by running the following commands in your terminal:
curl -L https://cloud.uol.de/public.php/dav/files/TN2bxBCbAS5DRDJ -o UNDATIPELAGIBACTER-GENOMES.db
curl -L https://cloud.uol.de/public.php/dav/files/ctRp8xRWwaPSnp5 -o UNDATIPELAGIBACTER-PAN.db
Now we have all the contigs-db files ready, we are ready to compute a conventional pangenome. Which is a necessary step prior to generating a pangenome graph. The topic of pangenomics is covered extensively in another tutorial, so here we will employ a pragmatic approach and use only what we need from it.
Generating an anvi’o pan-db is also straightforward: (1) put genomes of interest in a genomes-storage-db and (2) compute a pangenome from it.
We will use the program anvi-gen-genomes-storage for the first step, and for that, we first need to describe which genomes we are interested in using an external-genomes artifact. Since we are interested in all genomes now under CONTIGS-DBs, generating a file that describes all of them is as simple as running the following command:
anvi-script-gen-genomes-file --input-dir CONTIGS-DBs/ \
-o external-genomes.txt
This will create a two-column TAB-delimited file as explained here. Now we can complete the first step of computing a pangenome, and generate a genomes-storage-db for ourselves:
anvi-gen-genomes-storage -e external-genomes.txt \
-o UNDATIPELAGIBACTER-GENOMES.db
Once it is ready, we can generate the pangenome:
anvi-pan-genome -g UNDATIPELAGIBACTER-GENOMES.db \
--project-name UNDATIPELAGIBACTER \
--num-threads 8
And visualize it using anvi-display-pan:
anvi-display-pan -p UNDATIPELAGIBACTER-PAN.db \
-g UNDATIPELAGIBACTER-GENOMES.db
Which will give us the following interactive interface to play with this pangenome:
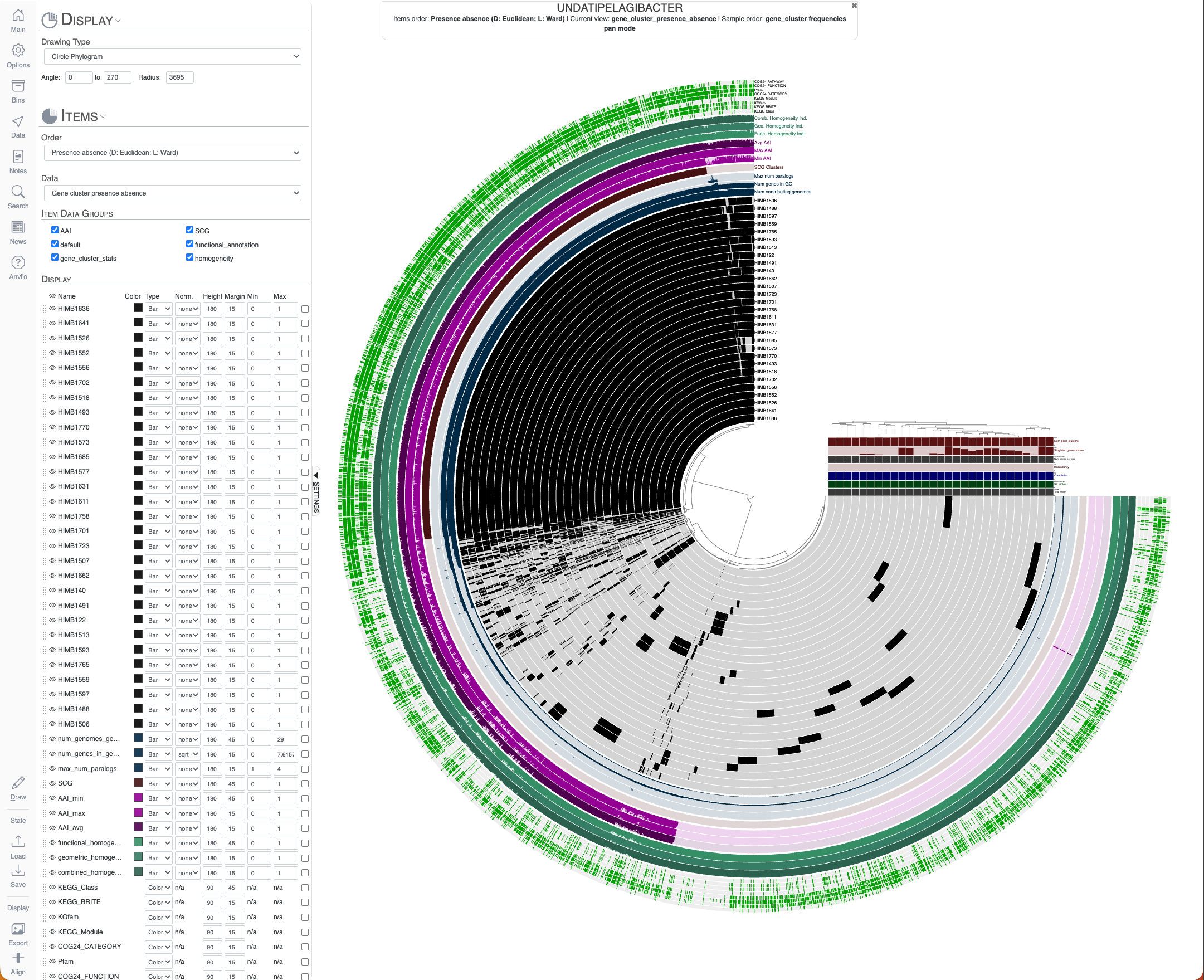
Undatipelagibacter pangenome with 29 complete genomes.
This interactive display and the pan-db itself offer a wide range of downstream analysis opportunities. But we will not go through them here since what to do with pangenomes is extensively covered elsewhere.
Instead, we will quickly move on to the primary purpose of this tutorial: turning a pangenome into a pangenome graph.
Computing the pangenome graph
Download the Undatipelagibacter pangenome graph instead?
If you want to skip this section, you can simply download the final products of the analysis steps listed below by running the following commands in your terminal:
curl -L https://cloud.uol.de/public.php/dav/files/8eZZYqNrAdXF4TA -o UNDATIPELAGIBACTER-PAN-GRAPH.db
As our paper also mentions and we reiterated at the beginning of this tutorial, pangenomics is indeed an extremely powerful tool to characterize the gene repertoire across a set of genomes and to quantify conserved and variable gene sets among closely related members of a taxon. The purpose of anvi-pan-genome-graph is to recover syntenic context of genes, and make them accessible for downstream exploratory or quantitative analyses, by turning pangenomes into pangenome graphs:
anvi-pan-genome-graph -e external-genomes.txt \
-g UNDATIPELAGIBACTER-GENOMES.db \
-p UNDATIPELAGIBACTER-PAN.db
The output messages from the program will walk you through a series of clearly labeled stages, each announced in the terminal with a green header, so you can follow the progress and make sense of what is going on. Without going too much in technical detail, here is a brief summary of what happens when you run the program, and what success (or failure) looks like:
Stage 1: Start-node selection. After displaying your settings, the program determines the first node in the graph. It will either use the gene you pointed to with the flag --start-gene, or it will automatically select the synteny gene cluster with the smallest average gene caller ID across the most genomes. The latter is a great choice that works well when you have reoriented your genomes so they all start at the same position like we did here in this tutorial.
Stage 2: SynGC contextualization. This is the most computationally intensive step. The algorithm splits conventional gene clusters calculated by anvi-pan-genome and described in pan-db into synteny gene clusters (SynGCs) by examining local genomic neighborhoods. At the end of this step, the program will print a simple summary table that will show the total number of genes across all genomes, number of gene clusters before contextualization, number of SynGCs after, and a breakdown of how many genes fell into core, rearrangement, accessory, singleton, duplication, and RNA categories.
When everything works well given your genomes (as in, the evolutionary or architectural distance between your genomes make them suitable for this kind of an analysis), the analysis should produce a SynGC count that is only modestly larger than the gene cluster count. If the SynGC count exceeds twice the gene cluster count, the program will stop and tell you that your data has too many repeats or multi-copy genes forcing over-splitting of the graph context. If things will fail, they will most likely fail at this stage. There is not a lot of ways to ameliorate this issue, but there are some tools in the program power users (or those who are really determined to figure things out) can try:
- Widen the context window by increasing
--min-k, or by lowering--alphaand/or--nparameters. - Account for inversions by using the
--inversion-awareflag if chromosomal inversions are common across the set of genomes considered. - Remove offending genomes from your analysis if there are clear offenders. Poorly assembled or poorly scaffolded genomes that contain chimeras or unrealistic inversions will ruin the graph for various reasons and have no place in a high-resolution analysis of genome architectures.
- Strip gene-level offenders by removing multi-copy genes with
--max-num-multi-copy-genesand--max-num-multi-copy-genes-per-genomeparameters. - Press the big red YOLO button by declaring the
--just-do-itflag, and bypass all the graph integrity checks and let the algorithm attempt to continue and see if the graph will magically converge (it likely won’t).
Stage 3: Graph connectivity check. Disconnected fragments (which are common with draft genomes assembled in many contigs) will be reported during this step with their sizes and the algorithm will automatically prune them to keep only the largest connected components. If the graph is still fragmented after pruning, the run will abort with a message about severe data inconsistencies. This is the second place where things can go horribly wrong if you have fragmented genomes that are fragmented in unlucky ways.
Stage 4: Edge directionality optimization. The program will optimize the directionality of edges throughout the graph to produce a directed acyclic graph that is suitable for downstream layout calculations, and will let you know how many edges had to be reversed. There may be zero reversals, which would be unusual but valid, or you may have many many reversals, which would indicate substantial amount of structural complexity in your pangenome. Which is what we are here to study, so all is well.
Stage 5: Genome distances and output. A successful run will end with a genome-distance matrix calculated from the graph topological properties, and a PAN-GRAPH.db file written to your disk with a final message summarizing the number of regions identified and whether they were classified as backbone or variable regions.
Here is the very end of a successful run in our case with the 29 Undatipelagibacter genomes:
(...)
COMPUTING GRAPH-BASED GENOME DISTANCES
===============================================
(...)
* d(HIMB1506,HIMB1491) = 0.303
* d(HIMB1506,HIMB140) = 0.31
* d(HIMB1506,HIMB1493) = 0.299
* d(HIMB1506,HIMB1488) = 0.284
* d(HIMB1506,HIMB122) = 0.311
* d(HIMB1506,HIMB1507) = 0.292
* d(HIMB1491,HIMB140) = 0.23
* d(HIMB1491,HIMB1493) = 0.225
* d(HIMB1491,HIMB1488) = 0.317
* d(HIMB1491,HIMB122) = 0.257
* d(HIMB1491,HIMB1507) = 0.252
* d(HIMB140,HIMB1493) = 0.244
* d(HIMB140,HIMB1488) = 0.313
* d(HIMB140,HIMB122) = 0.237
* d(HIMB140,HIMB1507) = 0.235
* d(HIMB1493,HIMB1488) = 0.299
* d(HIMB1493,HIMB122) = 0.253
* d(HIMB1493,HIMB1507) = 0.258
* d(HIMB1488,HIMB122) = 0.311
* d(HIMB1488,HIMB1507) = 0.286
* d(HIMB122,HIMB1507) = 0.237
* Done.
STORING PAN-GRAPH-DB
==============================================================
Pan Graph database ..........................................: A new database, ./UNDATIPELAGIBACTER-PAN-GRAPH.db, has been created.
✓ pan_genome_graph.py took 0:02:48.2
If everything went well, we have our pan-graph-db ready for visualization.
Interactive visualization of the graph
Interactive visualization of a pangenome graph is done through the program anvi-display-pan-graph. When you run it on the files we have just generated,
anvi-display-pan-graph -p UNDATIPELAGIBACTER-PAN-GRAPH.db \
-g UNDATIPELAGIBACTER-GENOMES.db
Your browser should pop-up with an interactive display that shows you something like this when you press the ‘Draw’ button:
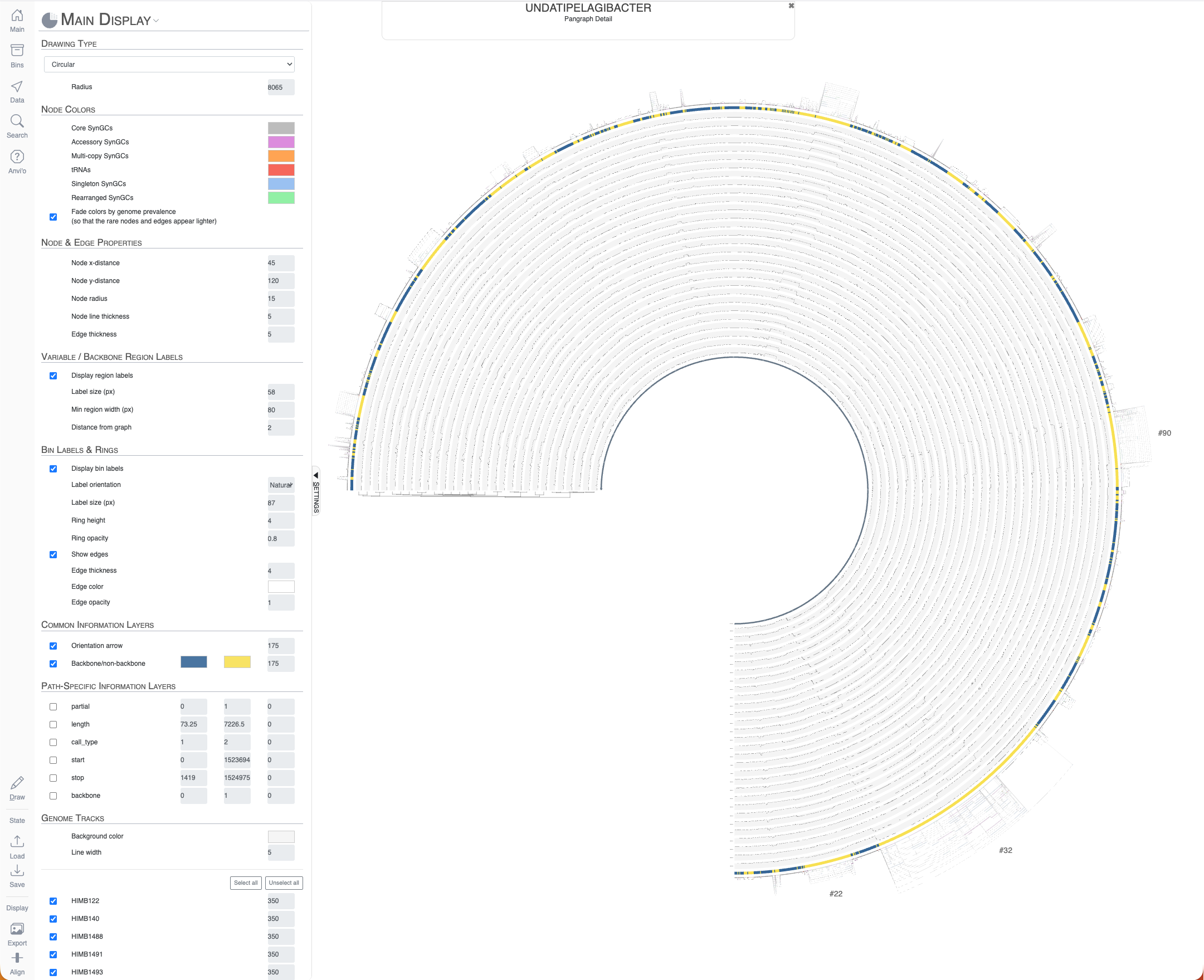
Undatipelagibacter pangenome graph with 29 complete genomes.
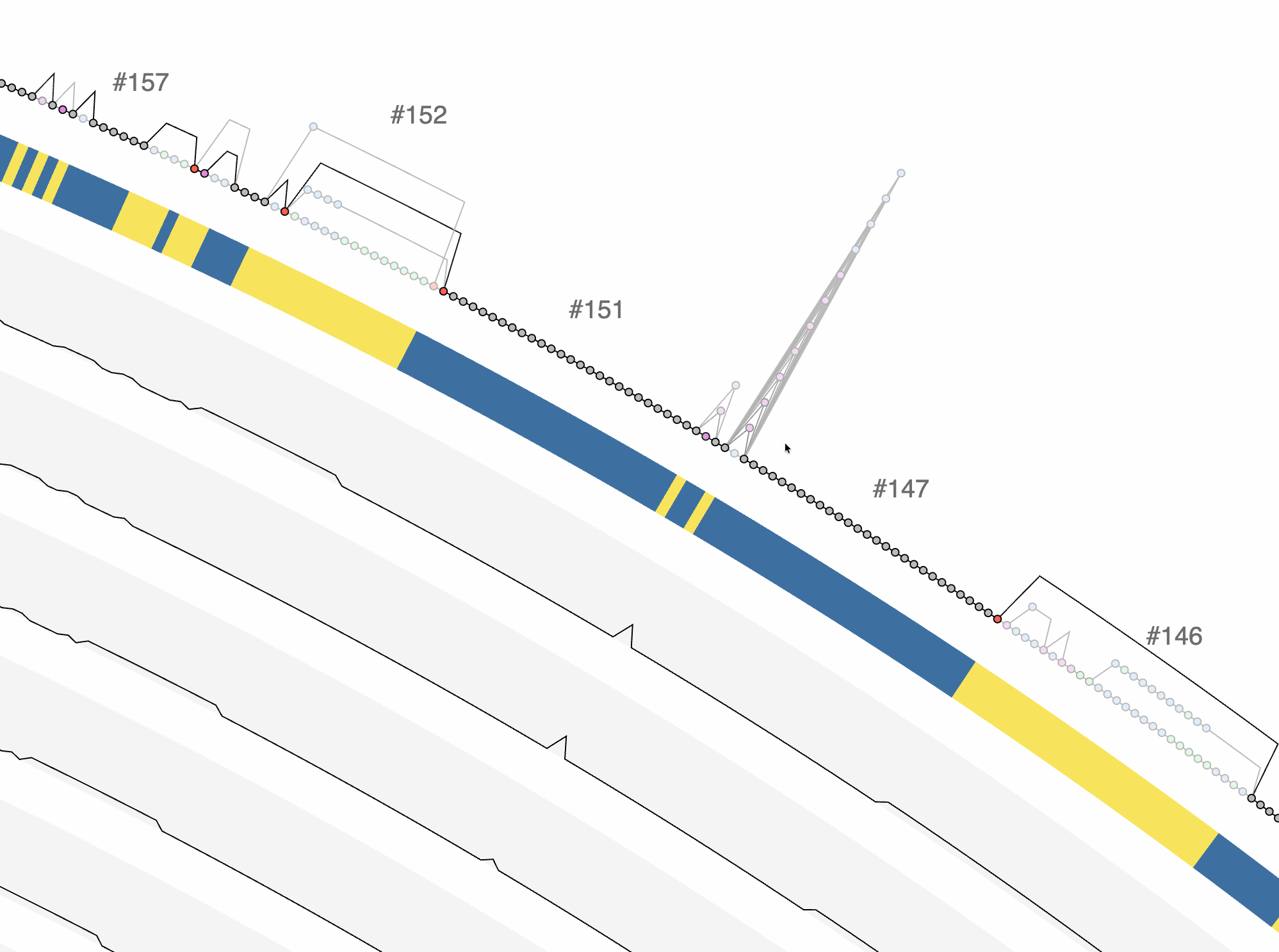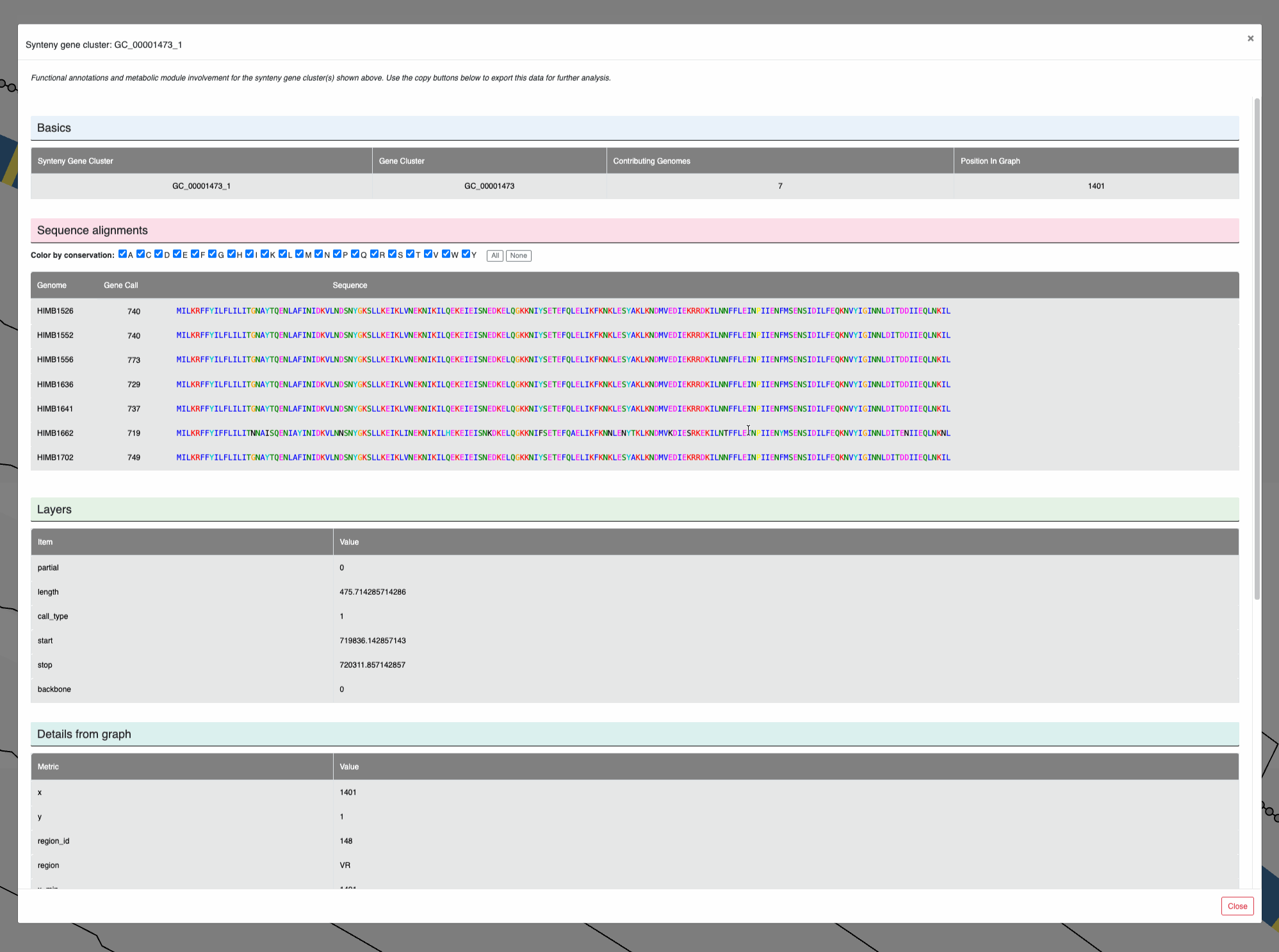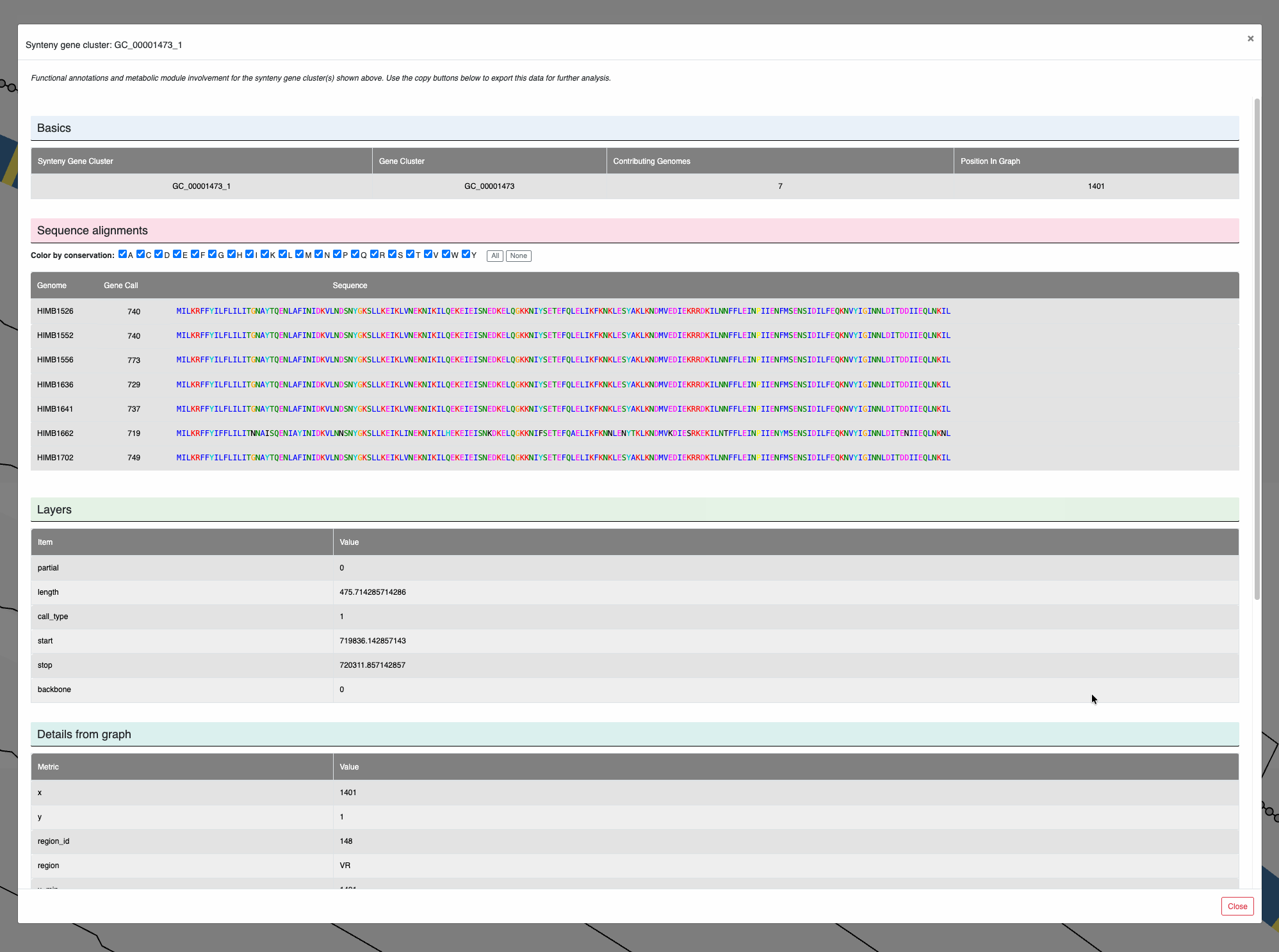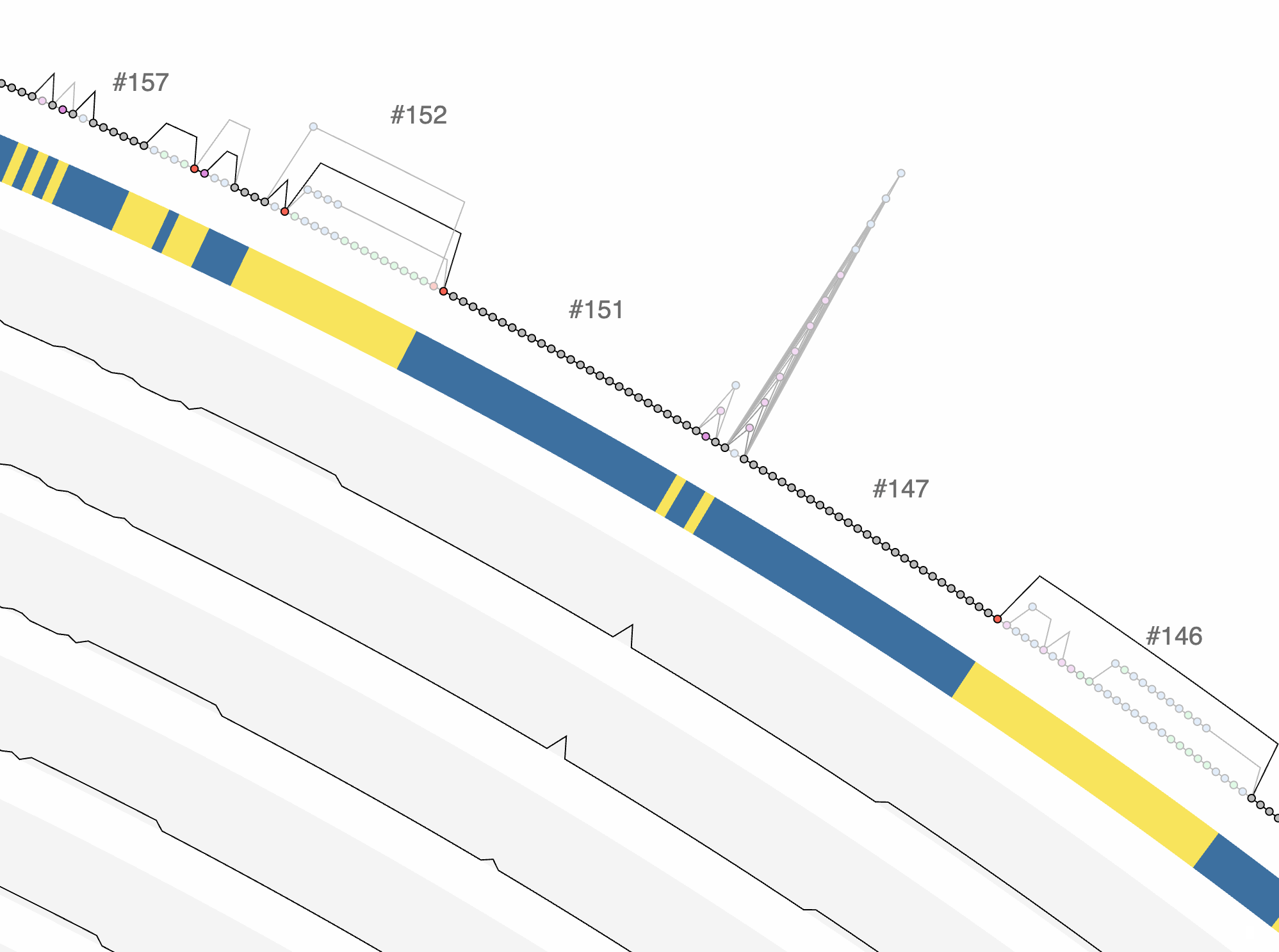
Before diving into the interface, it helps to understand the visual grammar of the graph. Each node represents a synteny gene cluster (SynGC), which describes a group of homologous genes that occupy the same syntenic position across one or more genomes. Edges connect SynGCs that are adjacent on at least one chromosome, so the paths through the graph trace the actual gene order of individual genomes. Where all genomes share the same gene neighborhood, the graph collapses into a single linear track. When genomes diverge (through gene INDELs or rearrangements), the graph branches into parallel paths that eventually/hopefully reconverge. The coloring distinguishes backbone regions (shown in blue by default), where all or nearly all genomes follow the same path, from variable regions (shown in yellow), where genomes take different routes through the graph. And this is exactly the kind of information a conventional pangenome cannot offer: the pangenome can tell you that a gene cluster is shared by 6 of 29 genomes, but only the pangenome graph can tell you that those 6 genomes carry it in a specific syntenic context between two backbone regions, forming a coherent variable island.
There are a lot of things one can do with the interactive interface, and it is difficult to cover all functionality it offers. One of the ways to explore the interface is through clicking on region IDs that are automatically assigned by anvi’o. In the screenshot above you already can see some regions numbered. How many of them are shown is a function of the size of the region and zoom level. By zooming into the graph with your mouse, you can see more of these region IDs to appear:
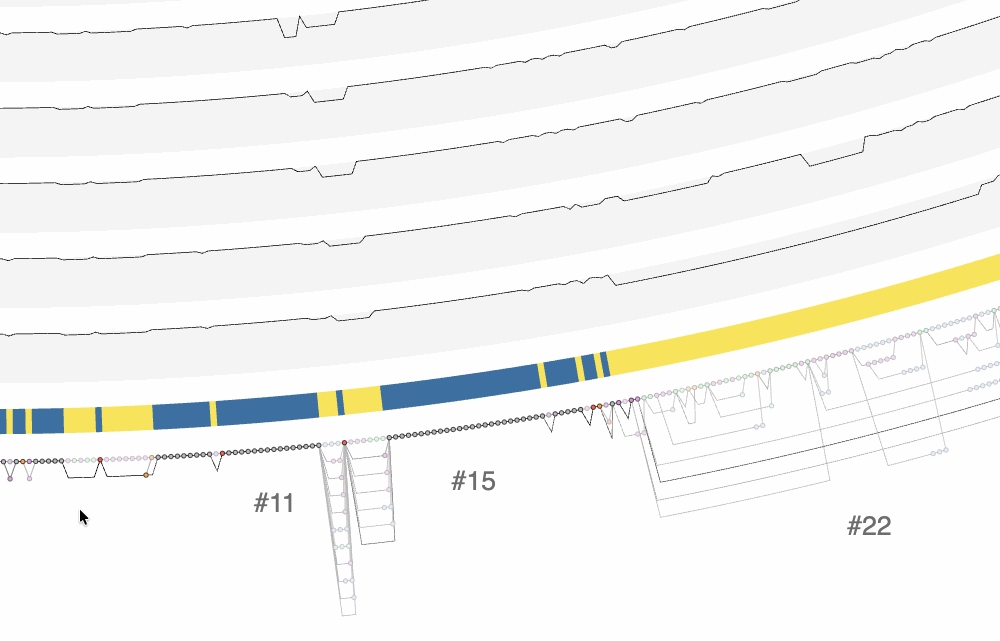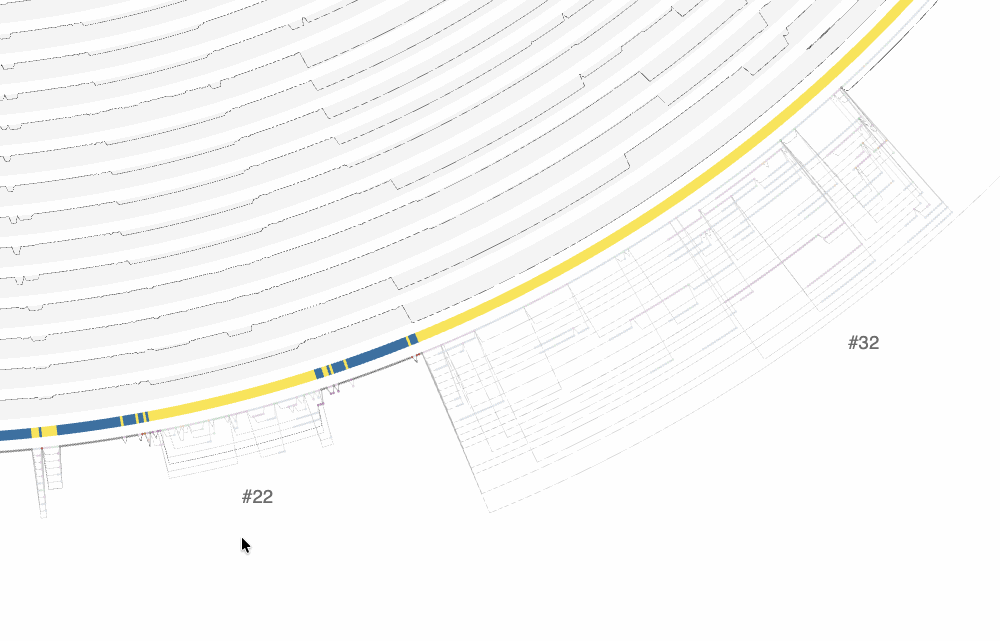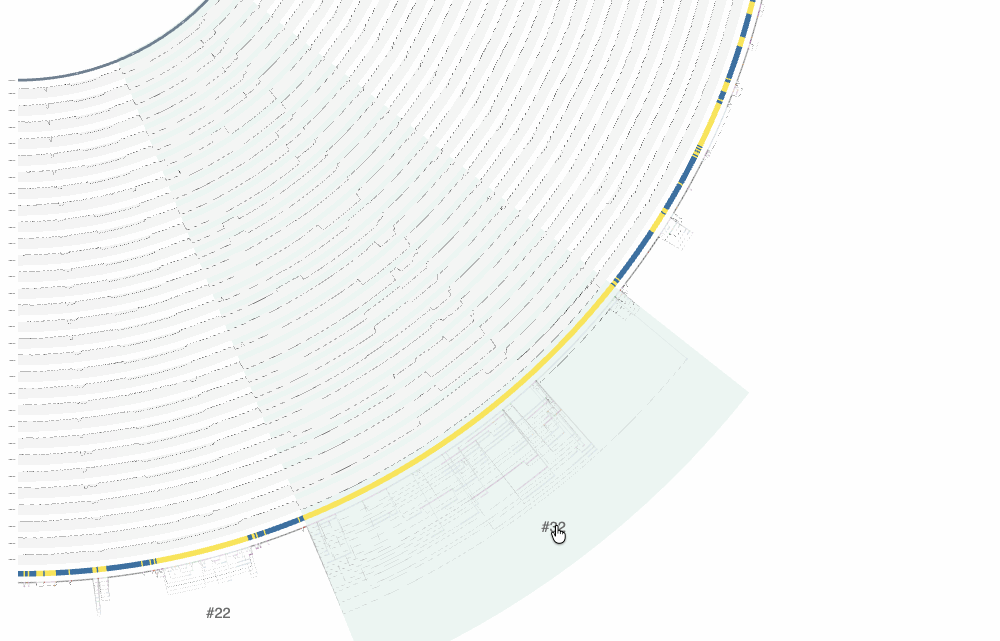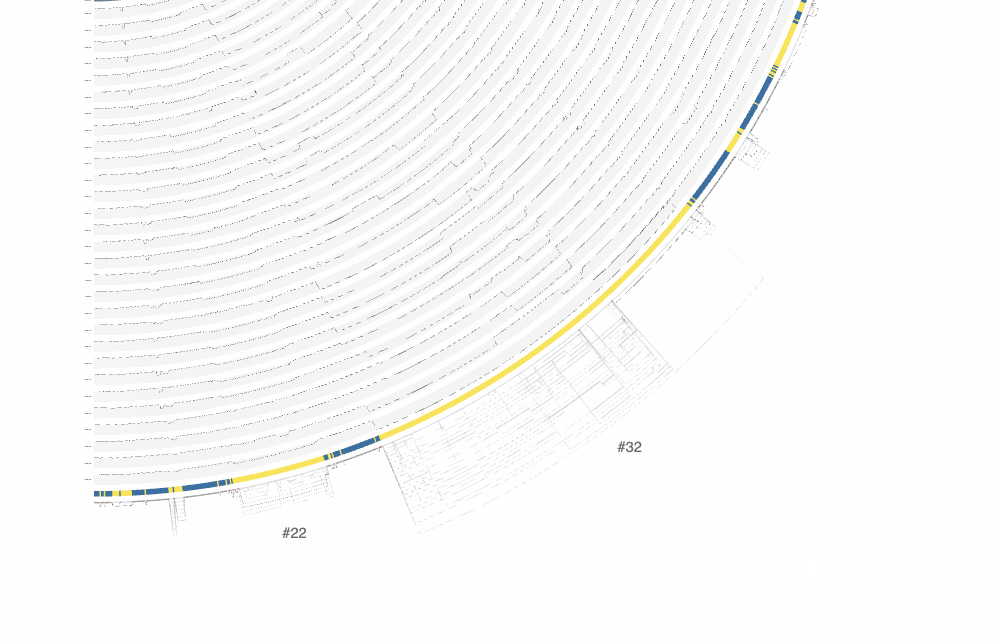
The effect of zooming in and zooming out on the interface.
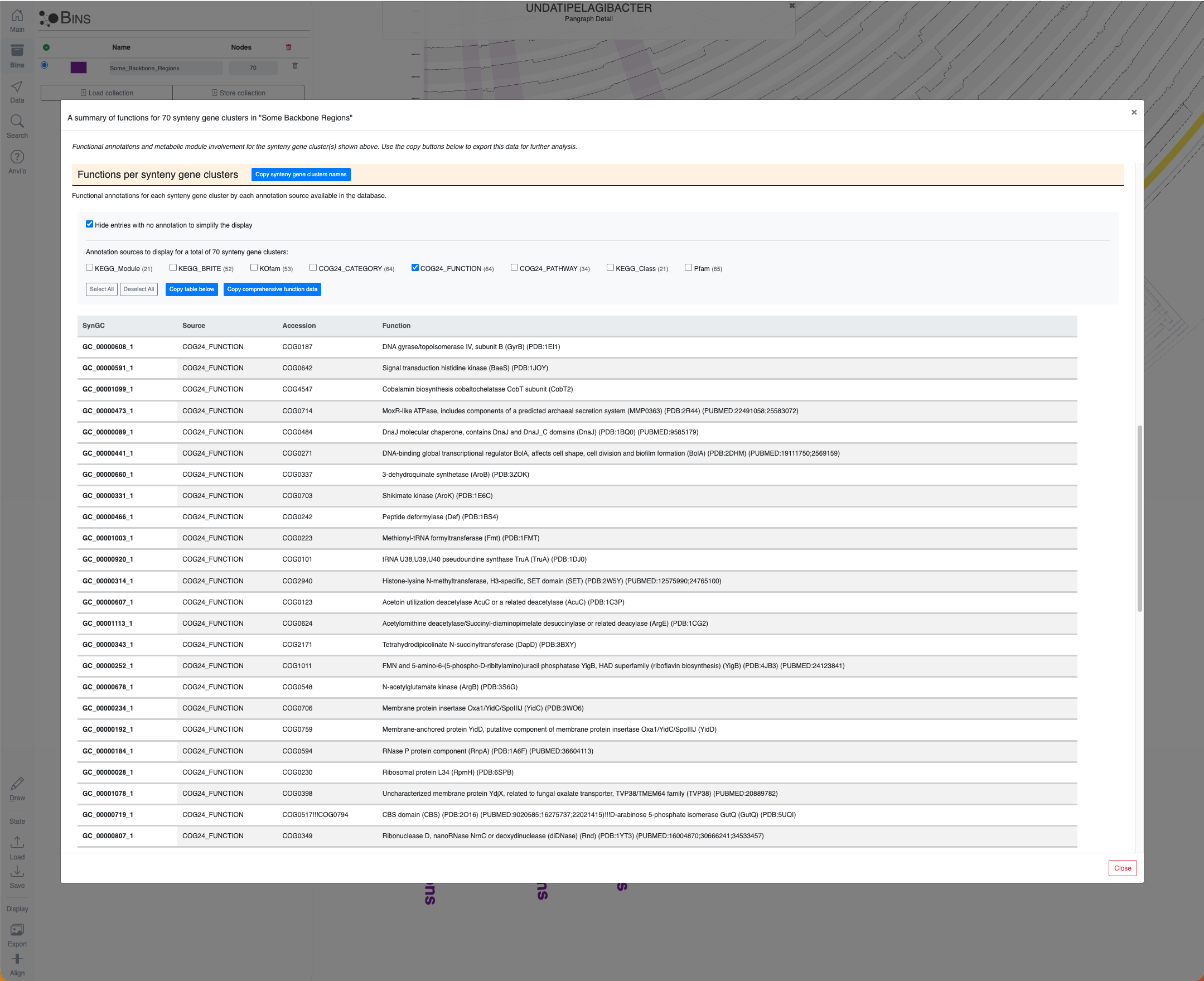
By clicking on these region IDs, or by putting them in bins entirely or manually by selecting nodes while clicking Alt, you can learn about the metabolic modules they are involved in, and the function annotation information for each SynGC. Here is the example output for region #32:
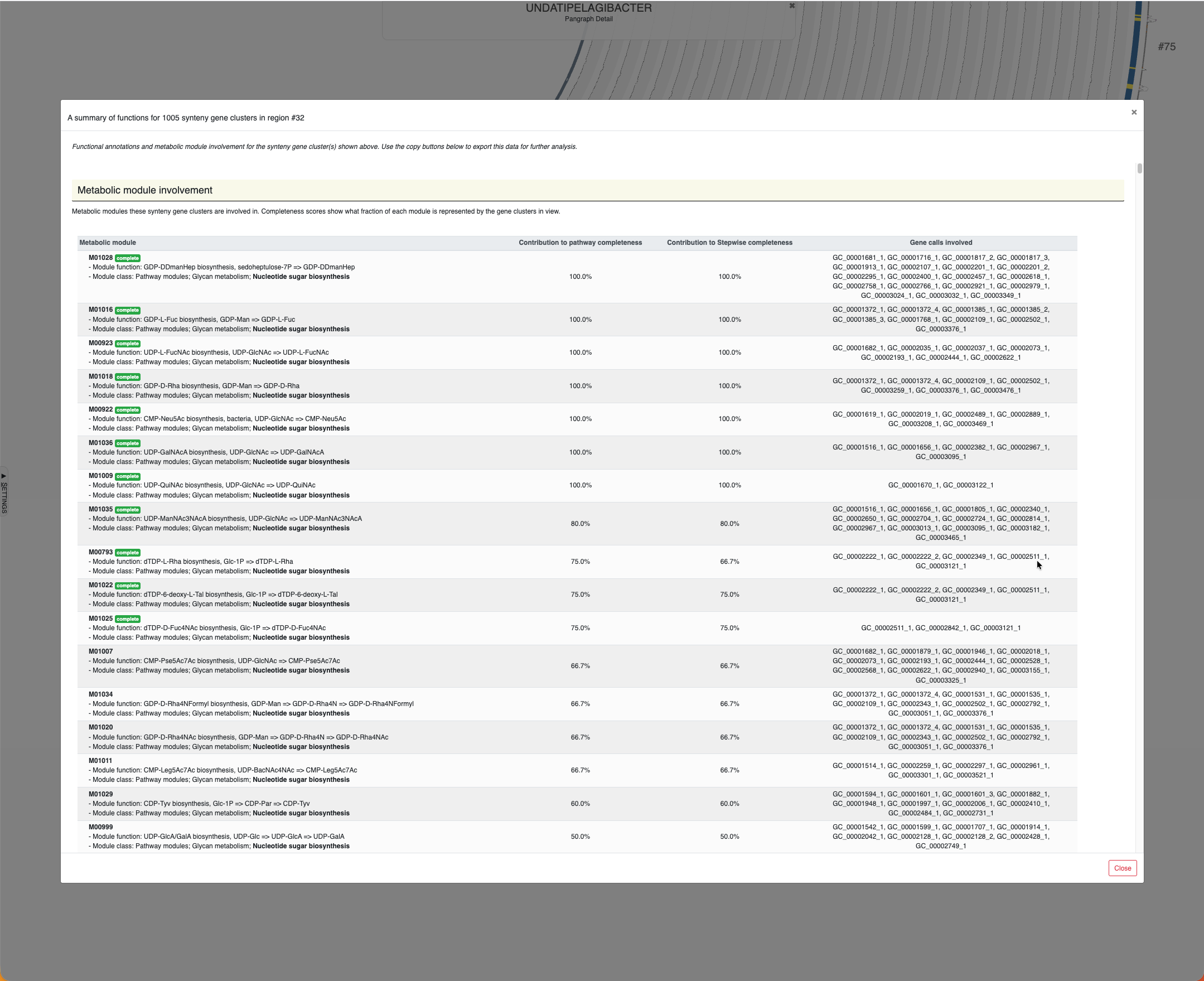
Region #32 in the Undatipelagibacter pangenome graph and the metabolic involvement of SynGCs encoded in this region.
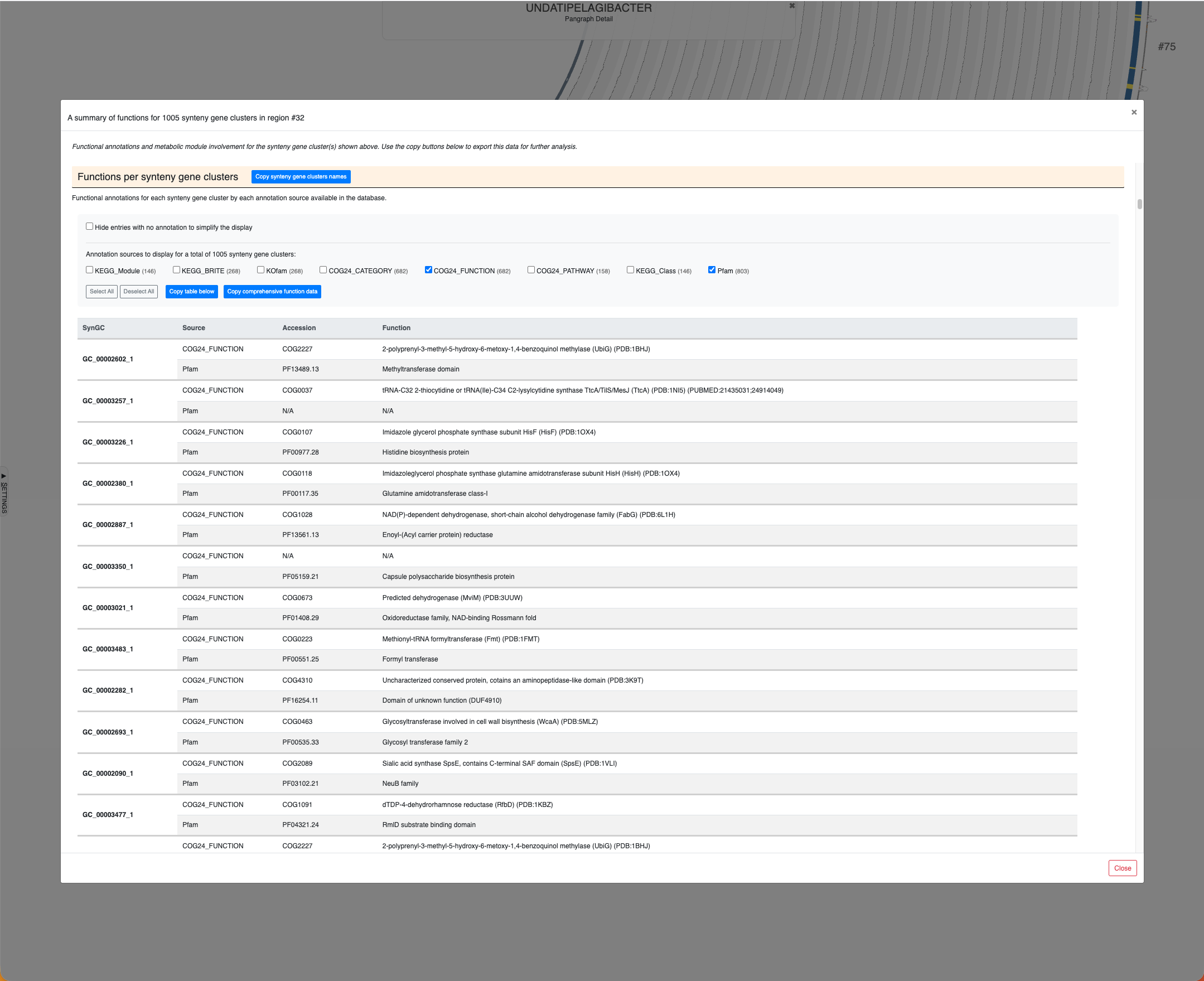
The same window as above, scrolled down to show the functions per SynGC section of it.
You can also arbitrarily add regions into any number of bins,

Some backbone regions added to a bin.
And show metabolic involvement of these SynGCs or their functional annotations by clicking the nodes button on the Bins panel:
Some exploratory insights into a bin.
You can also left-click on individual nodes to learn more about them, and see the amino acid sequence alignments among the genes that contribute to them:
Clicking on nodes.
The interactive interface is still evolving, and will tremendously benefit from any input you may have to share with us.
Summarizing the graph
Download the graph summary output instead?
If you want to skip this section, you can simply download the summary files generated in this chapter by running the steps listed below. For that, please run the following lines in your terminal:
curl -L https://cloud.uol.de/public.php/dav/files/8snz92oDqJeARDK -o UNDATIPELAGIBACTER-PAN-GRAPH-SUMMARY.tar.gz
tar -xzf UNDATIPELAGIBACTER-PAN-GRAPH-SUMMARY.tar.gz && rm UNDATIPELAGIBACTER-PAN-GRAPH-SUMMARY.tar.gz
Browsing the graph and gaining metabolic and functional insights into backbone and variable regions is one of the most important activities one can do with pangenome graphs to wrap their minds around the nature of the genomes they are studying. But beyond these exploratory and largely qualitative insights, most research questions will also need text based summaries of this display so they can be fed into LLMs, or statistical analysis or visualization tools to put numbers and/or significance values on various claims.
To generate text summaries of a given pan-graph-db, one can use the program anvi-summarize. When you run it without any collection,
anvi-summarize -p UNDATIPELAGIBACTER-PAN-GRAPH.db \
-g UNDATIPELAGIBACTER-GENOMES.db \
-o UNDATIPELAGIBACTER-PAN-GRAPH-SUMMARY
This will generate an output directory with multiple output files.
REGIONS.txt
One of the most powerful insights pangenome graphs offer is the precise delineation of backbone and variable regions among a given set of genomes, what is shown in blue and yellow in default in the interactive display of the graph.
The REGIONS.txt file simply describe these regions by giving each independent region a unique identifier.
region_id |
region_type |
x_min |
x_max |
num_synteny_gene_clusters |
num_gene_clusters |
num_gene_calls |
max_expansion |
min_expansion |
complexity |
complexity_normalized |
diversity |
diversity_normalized |
weight |
weight_normalized |
composite_variability_score |
complexity_mm_scaled |
diversity_mm_scaled |
expansion_mm_scaled |
weight_mm_scaled |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | backbone | 0 | 3 | 4 | 4 | 116 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 2 | variable | 4 | 4 | 2 | 2 | 29 | 1 | 1 | 1 | 0.034482758620689655 | 175.75 | 0.20897740784780025 | 29 | 1.0 | 0.1467011361576307 | 0.045454545454545456 | 0.8966836734693878 | 0.011363636363636364 | 1.0 |
| 3 | backbone | 5 | 6 | 2 | 2 | 58 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 4 | variable | 7 | 7 | 2 | 2 | 29 | 1 | 1 | 1 | 0.034482758620689655 | 139.75 | 0.16617122473246138 | 29 | 1.0 | 0.13853120058298232 | 0.045454545454545456 | 0.7130102040816327 | 0.011363636363636364 | 1.0 |
| 5 | backbone | 8 | 12 | 5 | 5 | 145 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 6 | variable | 13 | 17 | 5 | 5 | 10 | 5 | 0 | 0 | 0.0 | 196.0 | 0.23305588585017836 | 2 | 0.06896551724137931 | 0.0 | 0.0 | 1.0 | 0.056818181818181816 | 0.03571428571428571 |
| 7 | backbone | 18 | 18 | 1 | 1 | 29 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 8 | variable | 19 | 26 | 9 | 8 | 51 | 8 | 1 | 2 | 0.06896551724137931 | 132.88888888888889 | 0.1580129475492139 | 29 | 1.0 | 0.27359729994261633 | 0.09090909090909091 | 0.6780045351473923 | 0.09090909090909091 | 1.0 |
| 9 | backbone | 27 | 35 | 9 | 9 | 261 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 10 | variable | 36 | 36 | 1 | 1 | 4 | 1 | 0 | 0 | 0.0 | 0.0 | 0.0 | 4 | 0.13793103448275862 | 0.0 | 0.0 | 0.0 | 0.011363636363636364 | 0.10714285714285714 |
| 11 | backbone | 37 | 52 | 16 | 16 | 464 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 12 | variable | 53 | 55 | 18 | 18 | 28 | 3 | 0 | 9 | 0.3103448275862069 | 195.19753086419752 | 0.23210170138430147 | 19 | 0.6551724137931034 | 0.3073956966458033 | 0.4090909090909091 | 0.9959057697152935 | 0.03409090909090909 | 0.6428571428571428 |
| 13 | backbone | 56 | 56 | 1 | 1 | 29 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 14 | variable | 57 | 62 | 12 | 12 | 28 | 6 | 0 | 5 | 0.1724137931034483 | 192.77777777777777 | 0.22922446822565729 | 17 | 0.5862068965517241 | 0.30548837466205775 | 0.2272727272727273 | 0.9835600907029478 | 0.06818181818181818 | 0.5714285714285714 |
| 15 | backbone | 63 | 87 | 25 | 25 | 725 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 16 | variable | 88 | 88 | 1 | 1 | 10 | 1 | 0 | 0 | 0.0 | 0.0 | 0.0 | 10 | 0.3448275862068966 | 0.0 | 0.0 | 0.0 | 0.011363636363636364 | 0.3214285714285714 |
| 17 | backbone | 89 | 93 | 5 | 5 | 145 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| 18 | variable | 94 | 94 | 1 | 1 | 3 | 1 | 0 | 0 | 0.0 | 0.0 | 0.0 | 3 | 0.10344827586206896 | 0.0 | 0.0 | 0.0 | 0.011363636363636364 | 0.07142857142857142 |
| 19 | backbone | 95 | 96 | 2 | 2 | 58 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 | 29 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.0 |
| (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) |
For instance, it is obvious from the display that one of the most variable regions in the Undatipelagibacter pangenome graph is the region #32. Using this file, one can use the quantitative estimates about the regions anvi’o have identified in the graph to reach the same conclusion:
Disclaimer, the following BASH one-liner is a copy-paste from ChatGPT answering the prompt: “Read the attached REGIONS.txt file, a documentation for which is available at https://anvio.org/help/main/artifacts/pan-graph-summary/, and give me a single BASH one-liner that will recover the most variable region ID in this pangenome graph”.
awk -F'\t' 'NR>1 && $16>max {max=$16; id=$1} END{print id}' UNDATIPELAGIBACTER-PAN-GRAPH-SUMMARY/REGIONS.txt
When we run it in the terminal, this is what I get:
32
Excellent. Or we could ask it to give us a BASH one-liner that finds the variable region with the lowest max_expansion and highest complexity to identify ‘tower’ like structures:
awk -F'\t' 'NR>1 && $2=="variable"' UNDATIPELAGIBACTER-PAN-GRAPH-SUMMARY/REGIONS.txt | sort -t$'\t' -k8,8n -k10,10rn | head -1 | awk '{print $1}'
When I run this in my terminal, this is what I get:
148
Which is indeed a very intersting region. One can go much further than this, of course using additional files, such as SYNGCs.txt.
SYNGCs.txt
Regions are made of SynGCs, and each SynGC is described in the SYNGCs.txt file, which looks like this:
Please note that there are many columns that may be hidded as a function of the resolution of your screen, but you can view them by sliding the display below towards right.
node_id |
bin_name |
source_gene_cluster_id |
node_type |
region_id |
region_type |
node_x |
node_y |
num_genomes_present |
genomes_present |
partial |
length |
call_type |
start |
stop |
backbone |
COG24_CATEGORY_ACC |
COG24_CATEGORY |
COG24_FUNCTION_ACC |
COG24_FUNCTION |
COG24_PATHWAY_ACC |
COG24_PATHWAY |
KEGG_BRITE_ACC |
KEGG_BRITE |
KEGG_Class_ACC |
KEGG_Class |
KEGG_Module_ACC |
KEGG_Module |
KOfam_ACC |
KOfam |
Pfam_ACC |
Pfam |
Transfer_RNAs_ACC |
Transfer_RNAs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GC_00000001_1 | GC_00000001 | duplication | 65 | backbone | 723 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 477.0 | 1.0 | 362416.775862069 | 362893.775862069 | 1 | K | Transcription | COG1278 | Cold shock protein, CspA family (CspC) (PDB:1C9O) | ko00001 | KEGG Orthology (KO)»>09190 Not Included in Pathway or Brite»>09192 Unclassified: genetic information processing»>99973 Transcription | K03704 | cold shock protein | PF00313.29 | ‘Cold-shock’ DNA-binding domain | |||||||||
| GC_00000001_2 | GC_00000001 | duplication | 19 | backbone | 96 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 144.0 | 1.5 | 73906.1379310345 | 74050.1379310345 | 1 | K | Transcription | COG1278 | Cold shock protein, CspA family (CspC) (PDB:1C9O) | ko00001 | KEGG Orthology (KO)»>09190 Not Included in Pathway or Brite»>09192 Unclassified: genetic information processing»>99973 Transcription | K03704 | cold shock protein | PF00313.29 | ‘Cold-shock’ DNA-binding domain | |||||||||
| GC_00000002_1 | GC_00000002 | duplication | 75 | backbone | 798 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 354.0 | 1.0 | 429237.620689655 | 429591.620689655 | 1 | V | Defense mechanisms | COG2076 | Multidrug transporter EmrE and related cation transporters (EmrE) (PDB:2I68) | PF00893.26 | Small Multidrug Resistance protein | |||||||||||||
| GC_00000002_2 | GC_00000002 | duplication | 75 | backbone | 799 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 334.5 | 1.0 | 429581.120689655 | 429915.620689655 | 1 | V | Defense mechanisms | COG2076 | Multidrug transporter EmrE and related cation transporters (EmrE) (PDB:2I68) | ko02000 | Transporters»>Other transporters»>Electrochemical potential-driven transporters [TC:2] | K03297 | small multidrug resistance pump | PF00893.26 | Small Multidrug Resistance protein | |||||||||
| GC_00000003_1 | GC_00000003 | duplication | 63 | backbone | 715 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 1471.5 | 1.0 | 354607.844827586 | 356079.344827586 | 1 | E | Amino acid transport and metabolism | COG0665 | Glycine/D-amino acid oxidase (deaminating) (DadA) (PDB:3AWI) | ko00001 | KEGG Orthology (KO)»>09100 Metabolism»>09102 Energy metabolism»>00680 Methane metabolism [PATH:ko00680] | K22084 | methylglutamate dehydrogenase subunit A [EC:1.5.99.5] | PF01266.31 | FAD dependent oxidoreductase | |||||||||
| GC_00000003_2 | GC_00000003 | duplication | 123 | backbone | 1156 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 765.0 | 1.0 | 595759.568965517 | 596524.568965517 | 1 | E | Amino acid transport and metabolism | COG0665 | Glycine/D-amino acid oxidase (deaminating) (DadA) (PDB:3AWI) | ko01000 | KEGG Orthology (KO)»>09100 Metabolism»>09105 Amino acid metabolism»>00260 Glycine, serine and threonine metabolism [PATH:ko00260] | M00975 | Pathway modules; Amino acid metabolism; Serine and threonine metabolism | M00975 | Betaine degradation, bacteria, betaine => pyruvate | K00303 | sarcosine oxidase, subunit beta [EC:1.5.3.24 1.5.3.1] | PF01266.31 | FAD dependent oxidoreductase | |||||
| GC_00000004_1 | GC_00000004 | duplication | 213 | backbone | 1663 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 873.0 | 1.0 | 859511.75862069 | 860384.75862069 | 1 | I | Lipid transport and metabolism | COG1028 | NAD(P)-dependent dehydrogenase, short-chain alcohol dehydrogenase family (FabG) (PDB:6L1H) | COG1028 | Fatty acid biosynthesis | ko00001 | KEGG Orthology (KO)»>09100 Metabolism»>09101 Carbohydrate metabolism»>00040 Pentose and glucuronate interconversions [PATH:ko00040] | K22185 | D-xylose 1-dehydrogenase [EC:1.1.1.175] | PF13561.13 | Enoyl-(Acyl carrier protein) reductase | |||||||
| GC_00000004_2 | GC_00000004 | duplication | 215 | backbone | 1674 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 882.0 | 1.0 | 870404.568965517 | 871286.568965517 | 1 | I | Lipid transport and metabolism | COG1028 | NAD(P)-dependent dehydrogenase, short-chain alcohol dehydrogenase family (FabG) (PDB:6L1H) | COG1028 | Fatty acid biosynthesis | ko00001 | KEGG Orthology (KO)»>09100 Metabolism»>09101 Carbohydrate metabolism»>00040 Pentose and glucuronate interconversions [PATH:ko00040] | K22185 | D-xylose 1-dehydrogenase [EC:1.1.1.175] | PF13561.13 | Enoyl-(Acyl carrier protein) reductase | |||||||
| GC_00000005_1 | GC_00000005 | duplication | 179 | backbone | 1522 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 894.310344827586 | 1.0 | 806025.931034483 | 806920.24137931 | 1 | P | Inorganic ion transport and metabolism | COG0004 | Ammonia channel protein AmtB (AmtB) (PDB:1U77) (PUBMED:12023896;15876187;18362341) | ko02000 | Transporters»>Other transporters»>Pores ion channels [TC:1] | K03320 | ammonium transporter, Amt family | PF00909.27 | Ammonium Transporter Family | |||||||||
| GC_00000005_2 | GC_00000005 | duplication | 179 | backbone | 1523 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 1312.96551724138 | 1.0 | 807046.344827586 | 808359.310344828 | 1 | P | Inorganic ion transport and metabolism | COG0004 | Ammonia channel protein AmtB (AmtB) (PDB:1U77) (PUBMED:12023896;15876187;18362341) | ko02000 | Transporters»>Other transporters»>Pores ion channels [TC:1] | K03320 | ammonium transporter, Amt family | PF00909.27 | Ammonium Transporter Family | |||||||||
| GC_00000006_1 | GC_00000006 | duplication | 59 | backbone | 667 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 1197.0 | 1.0 | 307098.586206897 | 308295.586206897 | 1 | I | Lipid transport and metabolism | COG1012 | Acyl-CoA reductase or other NAD-dependent aldehyde dehydrogenase (AdhE) (PDB:1A4S) | COG1012 | Proline degradation | PF00171.28 | Aldehyde dehydrogenase family | |||||||||||
| GC_00000006_2 | GC_00000006 | duplication | 57 | backbone | 652 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 1230.6724137931 | 1.0 | 291393.965517241 | 292624.637931034 | 1 | I | Lipid transport and metabolism | COG1012 | Acyl-CoA reductase or other NAD-dependent aldehyde dehydrogenase (AdhE) (PDB:1A4S) | COG1012 | Proline degradation | PF00171.28 | Aldehyde dehydrogenase family | |||||||||||
| GC_00000007_1 | GC_00000007 | duplication | 67 | backbone | 743 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 613.5 | 1.0 | 380766.810344828 | 381380.310344828 | 1 | PF23169.2 | Halogenase D | |||||||||||||||||
| GC_00000007_2 | GC_00000007 | duplication | 182 | variable | 1593 | 0 | 15 | HIMB122,HIMB140,HIMB1488,HIMB1506,HIMB1507,HIMB1559,HIMB1573,HIMB1577,HIMB1597,HIMB1611,HIMB1631,HIMB1662,HIMB1723,HIMB1758,HIMB1765 | 0.0 | 869.8 | 1.0 | 830307.4 | 831177.2 | 0 | PF23169.2 | Halogenase D | |||||||||||||||||
| GC_00000008_1 | GC_00000008 | duplication | 311 | backbone | 2376 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 1002.0 | 1.0 | 1358314.5 | 1359316.5 | 1 | G | Carbohydrate transport and metabolism | COG2220 | L-ascorbate lactonase UlaG, metallo-beta-lactamase superfamily (UlaG) (PDB:2WYM) | ko01000 | Enzymes»>3. Hydrolases»>3.1 Acting on ester bonds»>3.1.4 Phosphoric-diester hydrolases»>3.1.4.54 N-acetylphosphatidylethanolamine-hydrolysing phospholipase D | K13985 | N-acyl-phosphatidylethanolamine-hydrolysing phospholipase D [EC:3.1.4.54] | PF12706.14 | Beta-lactamase superfamily domain | |||||||||
| GC_00000008_2 | GC_00000008 | duplication | 312 | variable | 2377 | 0 | 6 | HIMB1526,HIMB1552,HIMB1556,HIMB1636,HIMB1641,HIMB1702 | 0.0 | 460.5 | 1.0 | 1342273.66666667 | 1342734.16666667 | 0 | G | Carbohydrate transport and metabolism | COG2220 | L-ascorbate lactonase UlaG, metallo-beta-lactamase superfamily (UlaG) (PDB:2WYM) | PF12706.14 | Beta-lactamase superfamily domain | |||||||||||||
| GC_00000008_3 | GC_00000008 | duplication | 312 | variable | 2378 | 0 | 6 | HIMB1526,HIMB1552,HIMB1556,HIMB1636,HIMB1641,HIMB1702 | 0.0 | 223.5 | 1.0 | 1342691.66666667 | 1342915.16666667 | 0 | G | Carbohydrate transport and metabolism | COG2220 | L-ascorbate lactonase UlaG, metallo-beta-lactamase superfamily (UlaG) (PDB:2WYM) | |||||||||||||||
| GC_00000009_1 | GC_00000009 | duplication | 102 | variable | 993 | 0 | 11 | HIMB122,HIMB140,HIMB1488,HIMB1507,HIMB1513,HIMB1577,HIMB1611,HIMB1631,HIMB1723,HIMB1758,HIMB1765 | 0.0 | 869.454545454545 | 1.0 | 499033.181818182 | 499902.636363636 | 0 | G | Carbohydrate transport and metabolism | COG0483 | Archaeal fructose-1,6-bisphosphatase or related enzyme, inositol monophosphatase family (SuhB) (PDB:1AWB) | COG0483 | Gluconeogenesis | PF00459.31 | Inositol monophosphatase family | |||||||||||
| GC_00000009_2 | GC_00000009 | duplication | 75 | backbone | 795 | 0 | 29 | HIMB122,HIMB140,HIMB1488,HIMB1491,HIMB1493,HIMB1506,HIMB1507,HIMB1513,HIMB1518,HIMB1526,HIMB1552,HIMB1556,HIMB1559,HIMB1573,HIMB1577,HIMB1593,HIMB1597,HIMB1611,HIMB1631,HIMB1636,HIMB1641,HIMB1662,HIMB1685,HIMB1701,HIMB1702,HIMB1723,HIMB1758,HIMB1765,HIMB1770 | 0.0 | 955.396551724138 | 1.0 | 427109.120689655 | 428064.517241379 | 1 | G | Carbohydrate transport and metabolism | COG0483 | Archaeal fructose-1,6-bisphosphatase or related enzyme, inositol monophosphatase family (SuhB) (PDB:1AWB) | COG0483 | Gluconeogenesis | PF00459.31 | Inositol monophosphatase family | |||||||||||
| (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) | (…) |
Combining this file with the previous one, one can do a lot through the command line tools, Python, or R.
Because the summary files combine both structural information (region assignments, variability scores, genome membership) and functional annotations (COG, KEGG, Pfam) in flat tables, they are also well-suited for natural-language querying through large language models.
Just to demonstrate how these files are linked together and how accessible the output is, let’s upload both files to Claude and ask the following question:
The two file attached, REGIONS.txt and SYNGCs.txt are produced by the summary of a pangenome graph generated by anvi’o. The column names and their meanings are explained in this anvi’o artifact you should read:
https://anvio.org/help/main/artifacts/pan-graph-summary/
Please find the most variable region in this pangenome graph, and describe the functions encoded in this region by solely relying on the data in these files and very briefly describe the likely role of this region in the lifestyle of this organism.
Here is Claude’s response:
The most variable region appears to be the region 32.
Out of 341 regions in this pangenome graph (170 variable, 171 backbone), region 32 stands out dramatically with a composite variability score of 0.994 (on a 0–1 scale) — well above the runner-up, region 90, at 0.861. Here are its key statistics:
Metric Value Composite variability score 0.994 (next highest: 0.861) Synteny gene clusters 1,005 (721 found in only 1 of 29 genomes) Dominant COG category Cell wall/membrane/envelope biogenesis (53%) Key functions Sugar-nucleotide biosynthesis, glycosyltransferases, O-antigen ligases, LPS export, sialic acid-like sugar pathways This region appears to encode genome-specific surface polysaccharide biosynthesis machinery.
You may recall that region #32 was the same region we visually explored in the interactive display above. The LLM arrived at the same conclusion independently from the flat-text summaries alone, correctly identifying the surface polysaccharide biosynthesis island that is also visually striking in the graph. Something that is visible, is also quantifiable.
Final words
We hope our tutorial was useful for you to gain some insights into how to run pangenome graphs on your own datasets.
Please feel free to let us know if you have any questions.

If you are interested, you can continue with the reproducible bioinformatics workflow behind the Henoch et al. paper that starts with the data products generated in this tutorial, and the downstream analyses that led to the observations we described in our work:
https://merenlab.org/data/undatipelagibacter-pangenome-graph
Thank you for your time and interest.