



Table of Contents
Purpose and content check
The purpose of this post is to,
- Help the readers to appreciate the difficulties of binning complex metagenomes using short-read assemblies,
- Demonstrate what happens to our contigs across different binning algorithms and how this crucial information can be effectively visualized,
- Make suggestions for those who develop metagenomic binning algorithms and those who generate or use MAGs for their science to do better.
It contains about 3,000 words and will take little more than 15 minutes to read.

Introduction
If you are too cool for introductions and are only here to see some plots, jump here.
As the number of environmental metagenomes, binning algorithms, and metagenome-assembled genomes (MAGs) rapidly increase, it becomes easier to get washed away by the avalanche of data and forget what really matters. But when the dust settles from the current race of who is going to put together the largest number of genomes in one sit, perhaps we will once again realize that the convenience of outsourcing all decisions to some computer programs did not serve us as well as we thought it would.
The premise of metagenomic binning is relatively simple on paper: collect sample, sequence all DNA, assemble sequences, bin contigs, celebrate.

In reality, though, accurately reconstructing genomes from metagenomes using short-metagenomic reads is really difficult on many accounts of theory and practice (see “Accurate and Complete Genomes from Metagenomes” by Lin-Xing Chen et al. for a comprehensive overview, tips, and strategies).
The biggest problem of all is the poor assembly of short metagenomic reads. We all know and wish that long-read sequencing and Hi-C will soon start giving us near-complete genomes from metagenomes. But this is like how quantum computers will soon revolutionize computing (and in the process will likely make NP-complete problems disappear and render cryptography obsolete so we can all become a singularity of a broken family). We are not there yet, and until then we have to work with what we have. So if short reads are going to be around for a little longer, then what is the biggest risk?
The biggest risk of reconstructing genomes from metagenomes is to end up working with composite genomes, as composite genomes will influence ecological, pangenomic, and phylogenomic insights. Despite the community guidelines, avoiding composite genomes is not that easy even when we really really want to. Because counting the occurrence of single-copy core genes, which is the most commonly –and often the only– approach used to scrutinize new MAGs, is not bulletproof. Yes – single-copy core genes are not enough to detect contamination issues. So we will get bad bins, and in most cases we will not even know it unless we manually refine them and look at each contig. Most scientists in our field will agree that one of the most sorely missing pieces of our computational toolkit for genome-resolved metagenomics is the ability to estimate the quality and completion of a new genome without relying on any a priori information such as databases or gene families, and by using the entirety of the genomic data, and not only some parts of it. It is almost certain that multiple groups are pushing these boundaries, and we wish them well, as their solutions will continue to be useful even after the long-read sequencing revolution. But we are not there yet, either.
See this fresh blog post from Titus Brown for a database-based approach that uses GTDB and k-mers to look for contamination in genomes).
Long story short, in general we don’t have great assemblies and we don’t have bulletproof ways to identify contamination issues. What do we do, then?
The best we can do at the moment is perhaps to better understand what is going on within our genome bins that magically emerge from binning algorithms. And that is the purpose of this post. We will get there (or hopefully somewhere close) by discussing these two questions:
- Can we actually see the outcomes of the decisions computers make on our behalf during the binning process?
- Are there any scalable ways to deal with poor genome bins?
For those of you with little patience, answers to these questions are “sort of” and “no”. Feel free to go back to your work with a peaceful mind in your happy new year.
But if you are interested in what it means to sort of see the decisions of binning algorithms, and what turns that no into a yes easily but in a non-scalable fashion, then stick around to see some disturbing automatic binning decisions and mini manual refinement improvements.
Comparing metagenomic binning algorithms is difficult
It is difficult to compare binning algorithms since simulated or biological mock communities do not represent the complexity of environmental metagenomes, and metagenomes of non-simple habitats contain more organisms than we know about preventing benchmarks with a ground-truth.
So even when every publication introducing a new binning algorithm does so by showing that it is superior to all others, the question ‘which binning algorithm should we use’ still manages to roam without a single answer. This is because binning algorithms have different strengths and different weaknesses. Different binning algorithms may have different levels of success in dealing with metagenomes from different environments or constructed using different sequencing platforms. Moreover, different binning algorithms may have different levels of success in reconstructing different microbial populations even in a single metagenome.
Recognizing this mess, Christian M. K. Sieber and colleagues recently introduced DAS Tool, an approach that enables you to run multiple binning algorithms on the same dataset and aggregate the results to get your best final bins. This makes a lot of sense.
But most binning algorithms are going to agree when the input material is good (long contigs, lots of coverage, large number of samples, etc). When they disagree, however, it will largely be due to poor input material. And when the input material is not ideal, whether it is due to the limitations of sequencing chemistry or the peculiar biology of the study system, then it doesn’t really matter how great any individual tool or the one that will aggregate their results.
To establish this point better, the authors would like to offer you the Roomba vs dog poop challenge as an analogy. This is what happens when a state-of-the-art algorithm runs into a situation for which it was not prepared. Sadly, this is the case way too often when we are dealing with relatively complex metagenomes and short-read assembly: the data are not ideal for the vast majority of projects, and our algorithms have no idea what hit them. So when things are good, you are OK. And when things are bad, even DAS Tool will not be able to save a project while each Roomba is going haywire in their own way. So, the next question is, can we do better despite, especially when things are not ideal?
Whether we can do better or not requires us to understand what is really going on in our metagenomic bins. How do different binning algorithms compare to one another? How can we improve the final products? Doing that is relatively easy with simple metagenomes (as an example, here is how we did this before). But when things get complex, then we really have no idea what is going on in our bins beyond trivial summary tables that look good on paper, but say little in reality because MAGs are more than just a bunch of single-copy core genes to count.
One reason why it is difficult to compare different binning approaches at a high level of resolution is the scale of the problem. Hundreds of thousands of contigs end up in hundreds of genome bins across multiple binning algorithms. What makes this scale issue worse is the fact that there is no straightforward way to bring all the results into a single environment so we can actually start making sense of them.
Luckily, we partially solved this problem in the sixth version of anvi’o. Özcan Esen’s efforts gave us anvi-cluster-contigs, a program that enables anvi’o to run multiple binning algorithms on your data and seamlessly import the results into your profile database for comparative play time. Just to give you an idea, here is its help menu:

So this addresses the “but we don’t even get to play with data, so how can we even” part of the problem. Because we have everything in one place, and anvi’o is just rich with tools to play with data.
But if we get to play with data, which data to play with?
Something appropriately complex and realistic?
Some ocean metagenomes appear
Every corner of microbial ecology is filled with data. But the best data are the data that are in the progress of being analyzed so playing with it could end up being useful to its rightful owner as they go through it.
Enter a spontaneous thread in anvi’o Slack channel (which probably is not accessible anymore since we recently switched to Discord) by Jarrod Scott (who currently is a post-doctoral scientist at the Smithsonian Tropical Research Institute, Panama), which made it clear that he has been testing anvi-cluster-contigs on his ocean metagenomes.
The dataset contains 28 shallow water marine samples collected within a 20km radius around Isla Coiba in the Eastern Pacific Ocean. The data was co-assembled with MEGAHIT. Here is the output of anvi-display-contigs-stats for that assembly:
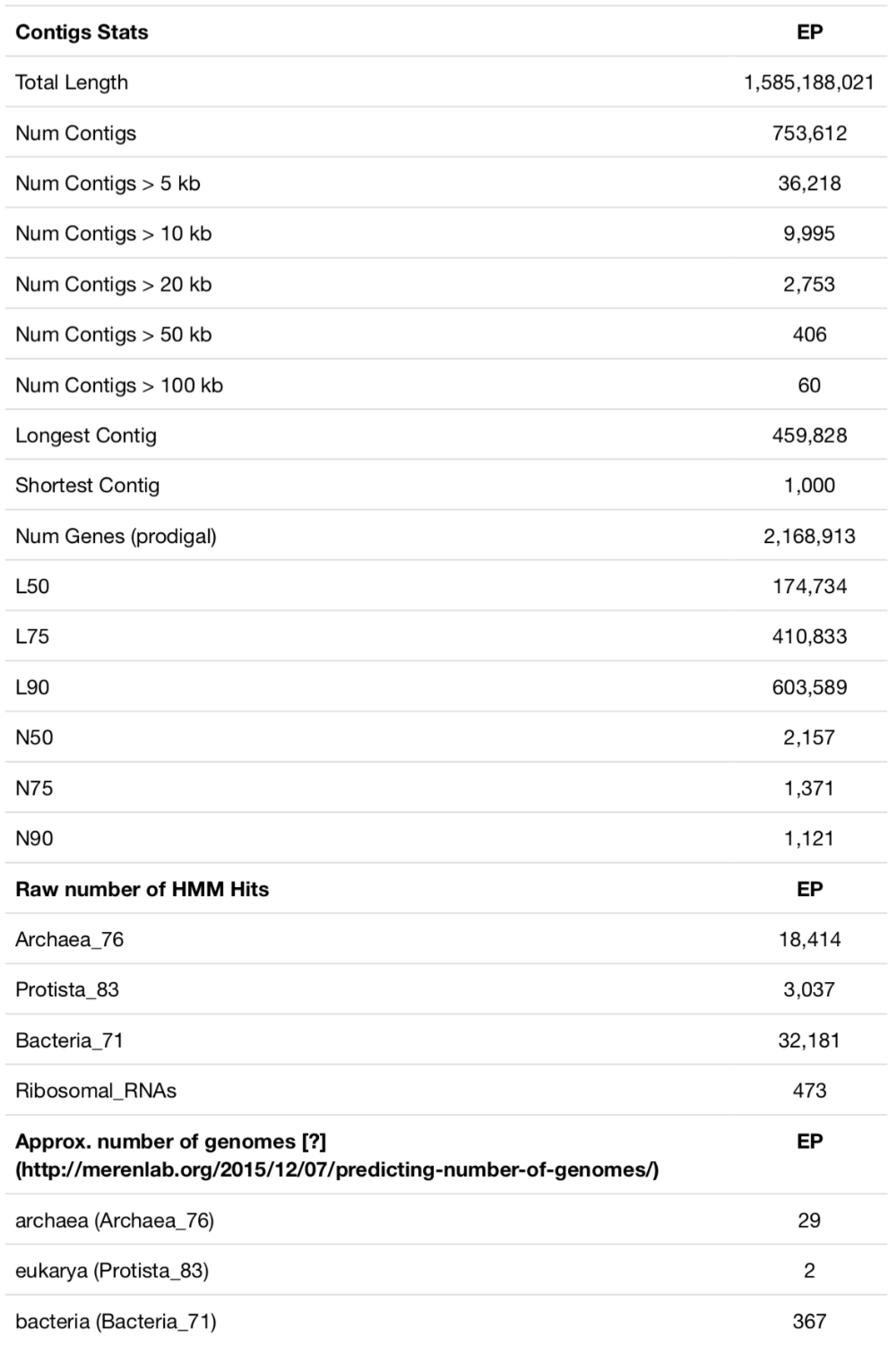
There are some good contigs, many bad contigs, and 2+ million genes that represent about 300 bacterial, 30 archaeal, and 2 eukaryotic genomes. With its size and complexity, this is a good representation of an ocean metagenome with short reads. Assembly statistics may improve slightly with a single-assembly strategy, by using IDBA or metaSPAdes, or may improve dramatically with the use of long-read sequencing in parallel. But this is what most researchers run into when they do metagenomic sequencing, so this assembly is not too far from reality.
Jarrod proceeds to map short reads from each metagenome back to the assembly, profiles each resulting BAM file with anvi-profile, merges the resulting single profile databases with anvi-merge, and runs anvi-cluster-contigs for CONCOCT, MaxBin 2.0, and MetaBAT 2, and also uses DAS Tool to aggregate results from each individual analysis.
Show/hide Important details of binning runs
Just to make sure it is clear for those of you who may be experts of these algorithms, these are the command lines anvi’o uses behind the scenes to run these algorithms (length threshold is by Jarrod):
# for CONCOCT
concoct --coverage_file contig_coverages.txt \
--composition_file contigs.fa \
--basename tmpnp2oj3a9 \
--threads 10 \
--length_threshold 1500
# metabat2
metabat2 -i contigs.fa \
-a contig_coverages.txt \
-o METABAT_ \
--cvExt \
-l \
--minContig 1500
# maxbin2
run_MaxBin.pl -contig contigs.fa \
-abund contig_coverages.txt \
-out MAXBIN_ \
-thread 10 \
-min_contig_length 1500
# dastool
DAS_Tool -c contigs.fa \
-i MAXBIN2.txt,METABAT2.txt,CONCOCT.txt \
-l MAXBIN2,METABAT2,CONCOCT \
-o OUTPUT \
--threads 10 \
--search_engine diamond
One thing to remember is that how each algorithm names bins, or else this can get confusing. CONCOCT adds the prefix Bin_, MaxBin adds the prefix MAXBIN_, and MetaBAT adds the prefix METABAT__ (with two underscores). DAS Tool then adds its own Bin_ prefix to the parent name. So DAS Tool Bin_Bin_13 is CONCOCT bin Bin_13 and Bin_METABAT__219 is MetaBAT bin METABAT__219.
So we have everything.
Visualizing contig fate across binning algorithms with alluvial diagrams
Each alluvial diagram you will see in this section aims to answer this question over and over again:
What do we see when we take all contigs binned together by an algorithm and ask “in which bins did these contigs appear in other algorithms?”.
Since all the binning results are in anvi’o, it was relatively easy to do this. We exported contig/bin associations for each binning algorithm using anvi-export-collection, and processed those output files to answer the question posed above using this program.
Let’s start.
Example 1
Here is one of our diagrams. We will talk about this first one extensively before we move on to the next to make sure it is abundantly clear to everyone what this shows. Please click on the image to see it full-screen first:
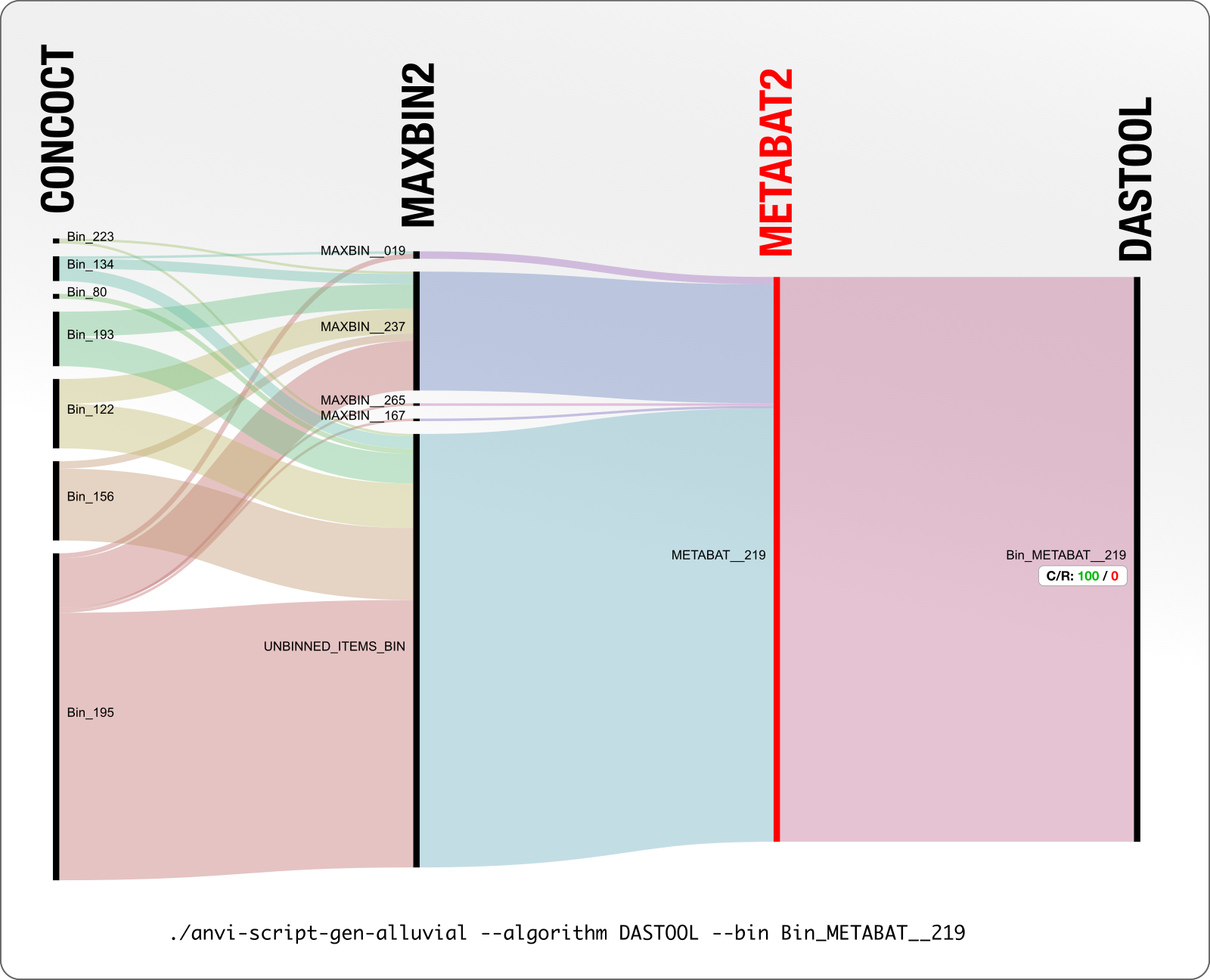
Here we take all contigs DAS Tool identified as Bin_METABAT__219 and see in what bins they appeared across other binning algorithms that work on the same data.
MetaBAT layer is shown in red to remind you that the basis of DAS Tool’s decision was the MetaBAT bin (as you can tell from the name DAS Tool gave to the final bin).
The C/R values indicate the completion and redundancy of a given bin. So according to this diagram we understand that the DAS Tool reported the MetaBAT bin METABAT__219 as is, which has a 100% completion and 0% redundancy according to anvi’o. Anvi’o determines this by counting the occurrence of bacterial single-copy core genes in this genome bin.
So what is up with MaxBin and CONCOCT? Before we move on, here is a critical point about our visualization approach. For instance, when you look at the size of MAXBIN_167, it looks too small to be a metagenomic bin. But what is shown here is not the entirety of MAXBIN_167, which in fact is 2.4 Mpb, but instead, only the amount of MAXBIN_167 contigs that ended up in METABAT_219.
Going back to “what is up with MaxBin and CONCOCT?” question: yes, this figure also shows that the set of contigs that were described in a single bin by METABAT, and chosen by DAS Tool as the final bin, were distributed across five bins in MAXBIN, and seven bins in CONCOCT. A great deal of confusion and disagreement here.
Example 2
In contrast, here is a better example. You can tell that the extent of agreement is much higher in this particular case.
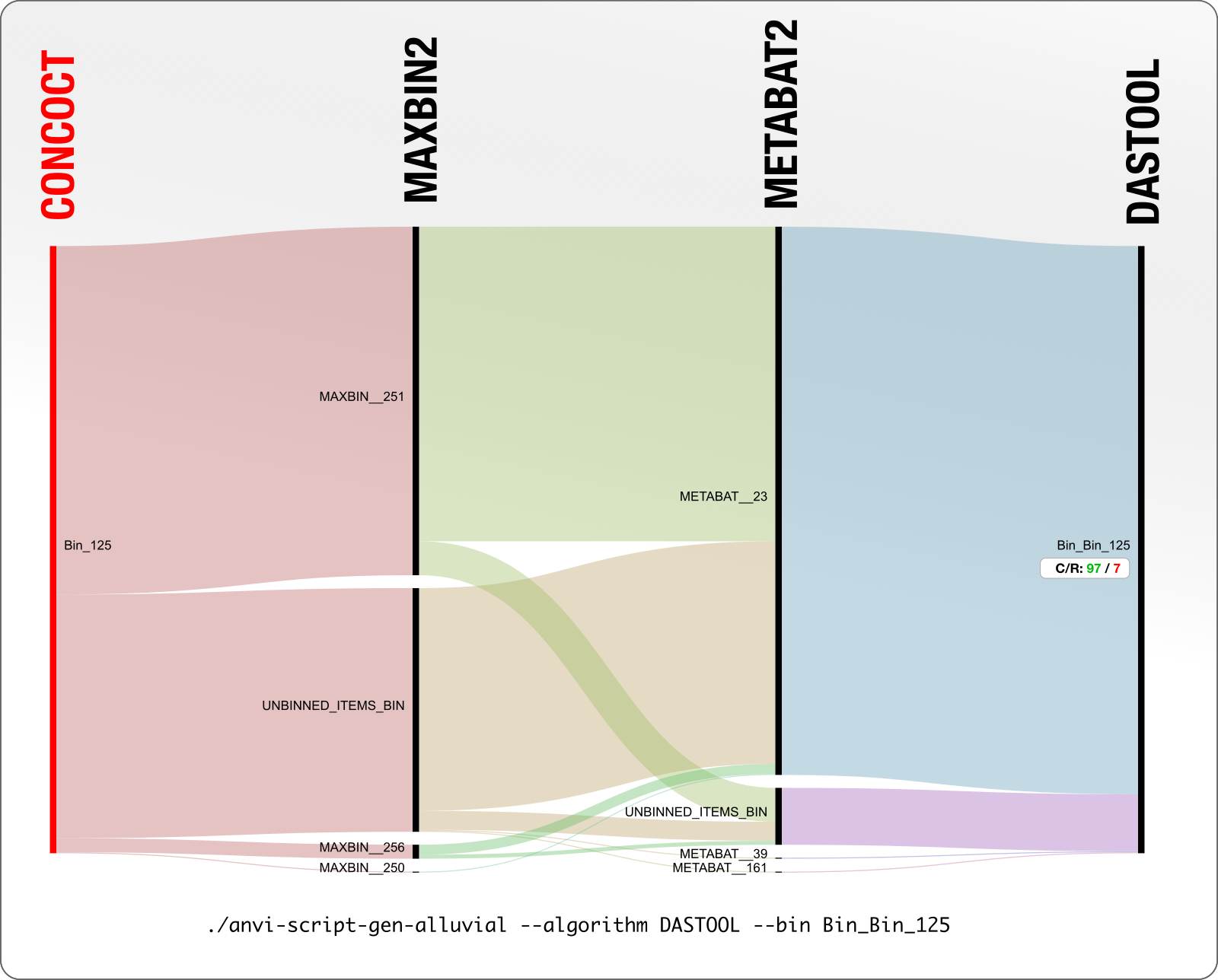
In this example DAS Tool chooses a CONCOCT bin as the final bin for these set of contigs, which is 97% complete and 7% redundant. Good for CONCOCT.
But then as you can see, MaxBin did not place half of these contigs in any bin, which is represented by UNBINNED_ITEMS_BIN, and split the rest of the contigs into three bins. MetaBAT, on the other hand, was largely in agreement with CONCOCT as it put most of these contigs in a single bin. Still, seeing contigs that were put together in a single bin by one algorithm to be distributing into multiple bins by other algorithms is not a happy place. If this makes you question a lot of things, you are not alone.
Example 3
This is a particularly disturbing example:
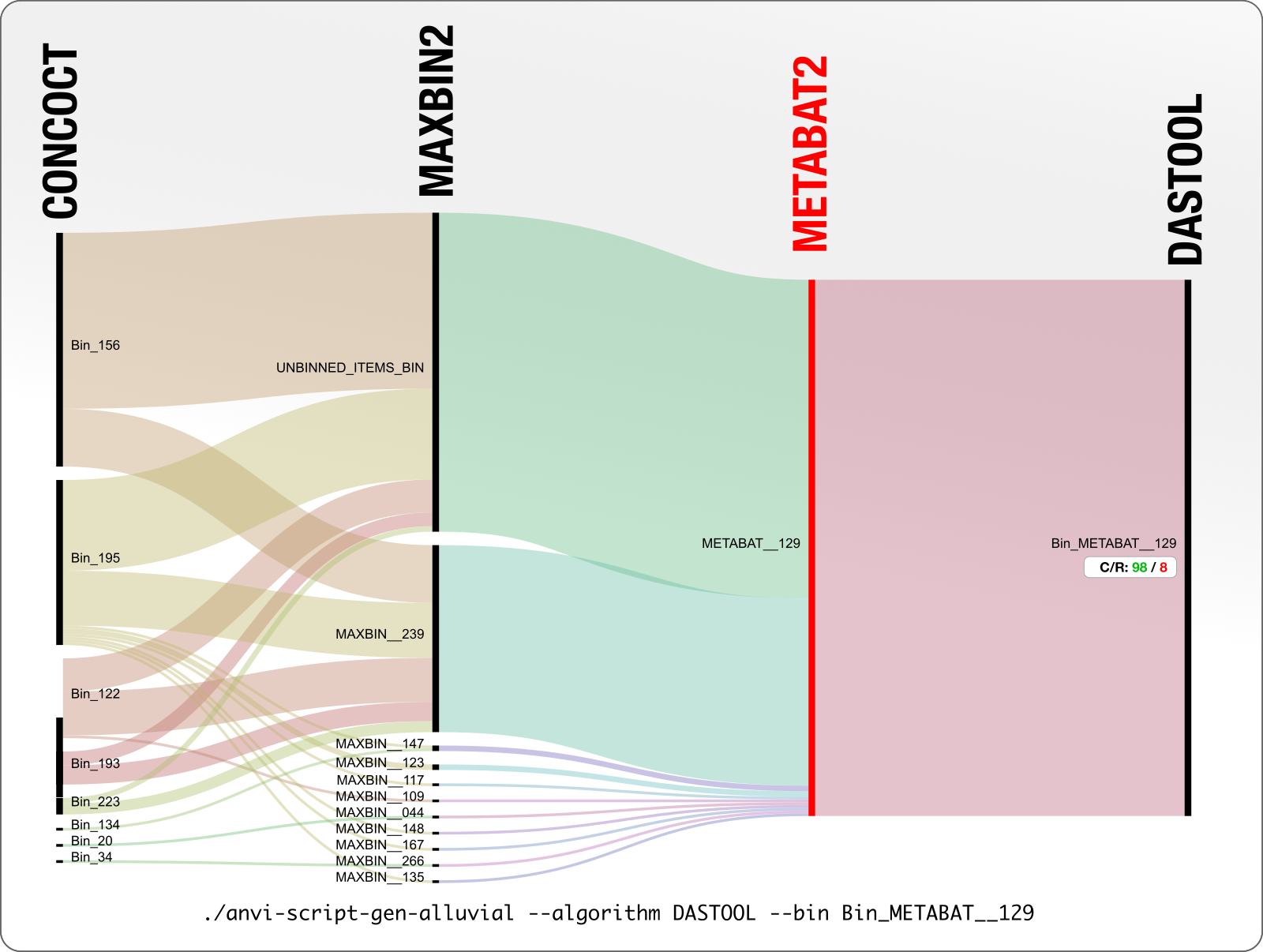
A 98% complete MetaBAT bin with only 8% redundancy, is split into 8 CONCOCT bins, and more than half of it is not binned by MaxBin.
Example 4
Here is another example:
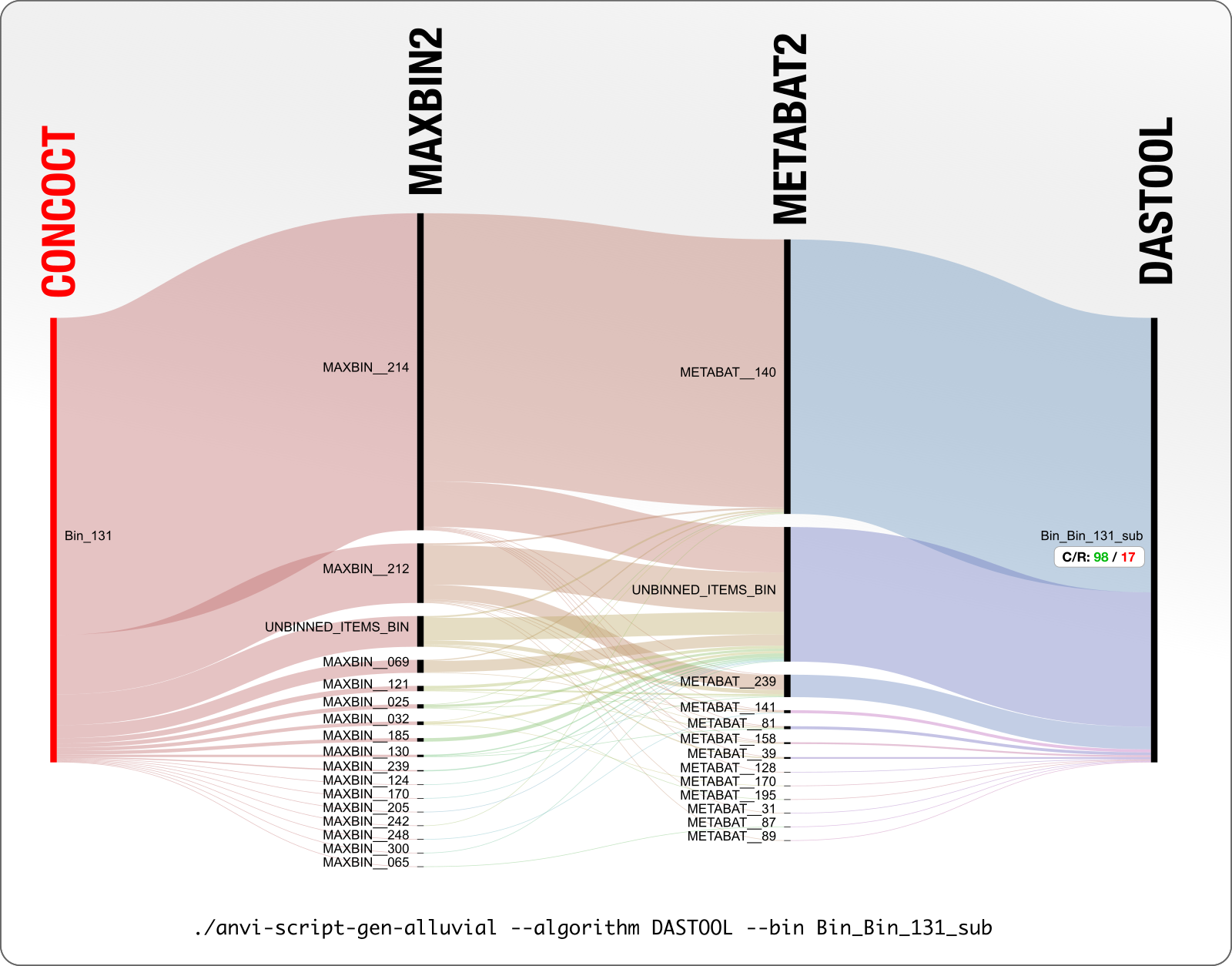
In this case DAS Tool reports only a part of a CONCOCT bin (you can tell by the _sub postfix in the name). This is where you watch your Roomba from your pet-cam approaching head-on towards some accident. The final bin has 98% complete and 17% redundant.
Similar to the case of MAXBIN_167 discussed above, CONCOCT Bin_131 shown here is only 60% of the bin that ended up being reported by DAS Tool. In this case it is hard to make sense of what led to this decision to split Bin_131 in this particular way. But seeing that these contigs ended up in 16 MaxBin and 12 MetaBAT bins is not encouraging.
Let’s turn the question around and say we will focus on the entirety of Bin_131 and see what is happening. Here is a diagram of the same bin, but starting with CONCOCT result, instead of DAS Tool:
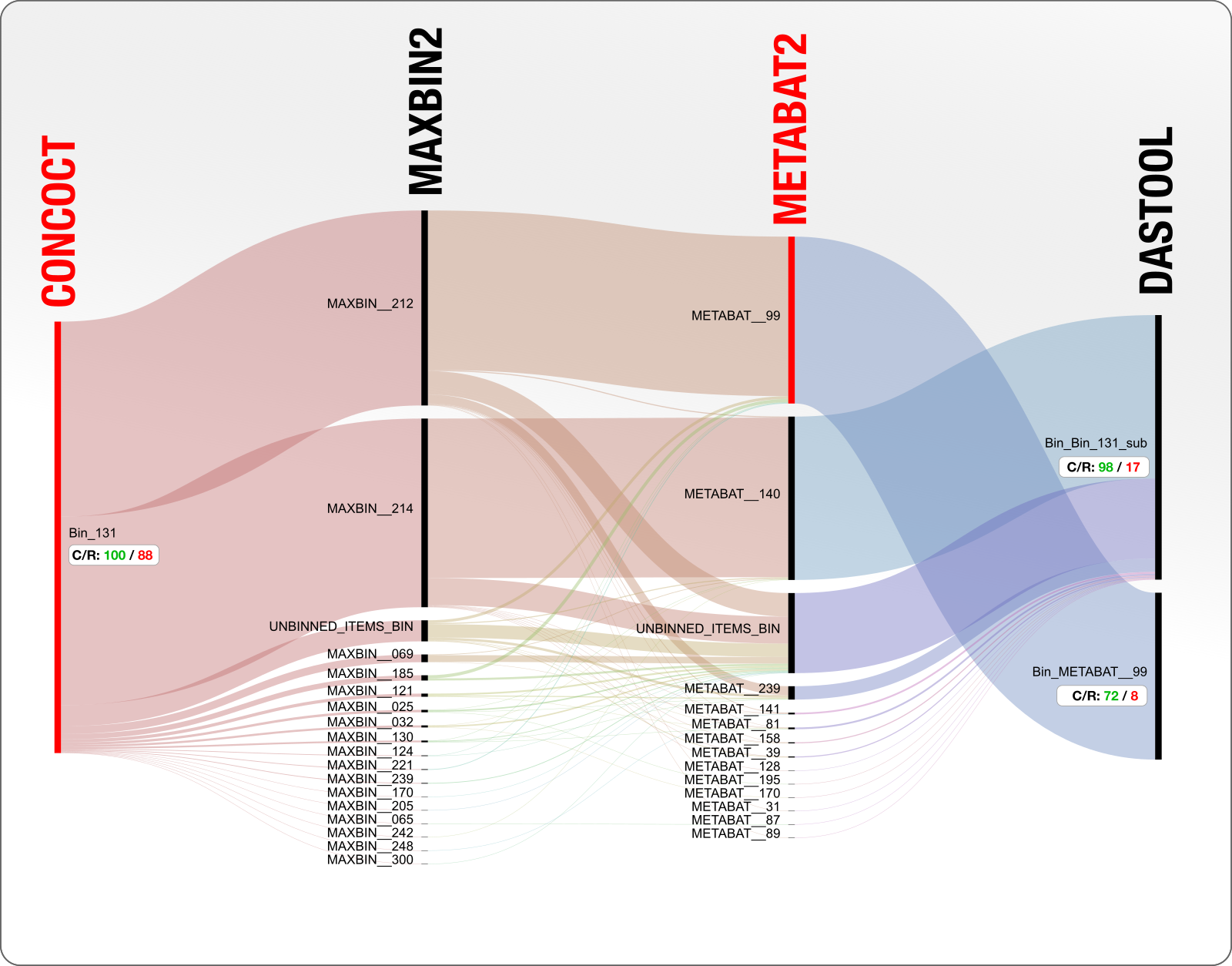
OK. So CONCOCT came up with a single bin that is 100% complete and 88% redundant (which likely contains two population genomes), everyone else put these contigs into two large bins (good job MetaBAT and MaxBin) and some random other bins (bad job everyone), and DAS Tool ended up deciding to use some of the contigs from the CONCOCT bin, and some others from the MetaBAT, reporting two genomes, one of which with 17% and the other with 8% redundancy.
#SAD indeed, but this looks like a clear win for the Roomba here. That said, the bin with 17% redundancy will not survive any quality criteria and will be eliminated from the analysis :/ Here is the key question: was there a way to save it?
OK. Let’s turn the question around one more time, and this time in such a way that the answer would help us see what was on the carpet here at the first place.
This time we are asking this: what if we manually refined the CONCOCT bin Bin_131 and add those results onto this display?
Here is our answer:

There is a lot to talk about here, but let’s first focus on the screenshot from the anvi’o interactive interface shown below.
Here, the contigs are organized based on sequence composition, as you can see they largely split into two major branches. The data shown here is detection, and the min/max values are set to 0.25 and 0.50. Which means, if less than 25% of a given contig has at least 1X coverage in a given metagenome, it will not have a black bar in the corresponding layer. In parallel, if more than 50% of it covered with at least one short read, then it will be shown with a full bar.
Since we are using only tetra-nucleotide frequency to organize these contigs, the detection signal has no influence on the way the center dendrogram organizes them. The difficulty here is that these two populations are co-occurring quite abundantly only in three metagenomes, so differential coverage signal is largely useless to them. But if you squint your eyes, you can tell that actually only one of them is detected faintly in the last two samples. So tetra-nucleotide frequency resolves distinct populations, and their ecology agrees with that resolution. It is a piece of cake for your eyes to pick up that signal and for your expertise to make sense of it, but for the Roomba it is just too much.
Now when you look at the diagram above, you see that now these two bins, which simply organize the same set of contigs differently, have a total redundancy of 9% and 4%. And to reach this outcome without sacrificing the completion of one of these bins, you would have had to start with the poorly done CONCOCT bin, rather than masterfully done DAS Tool sub-bin.
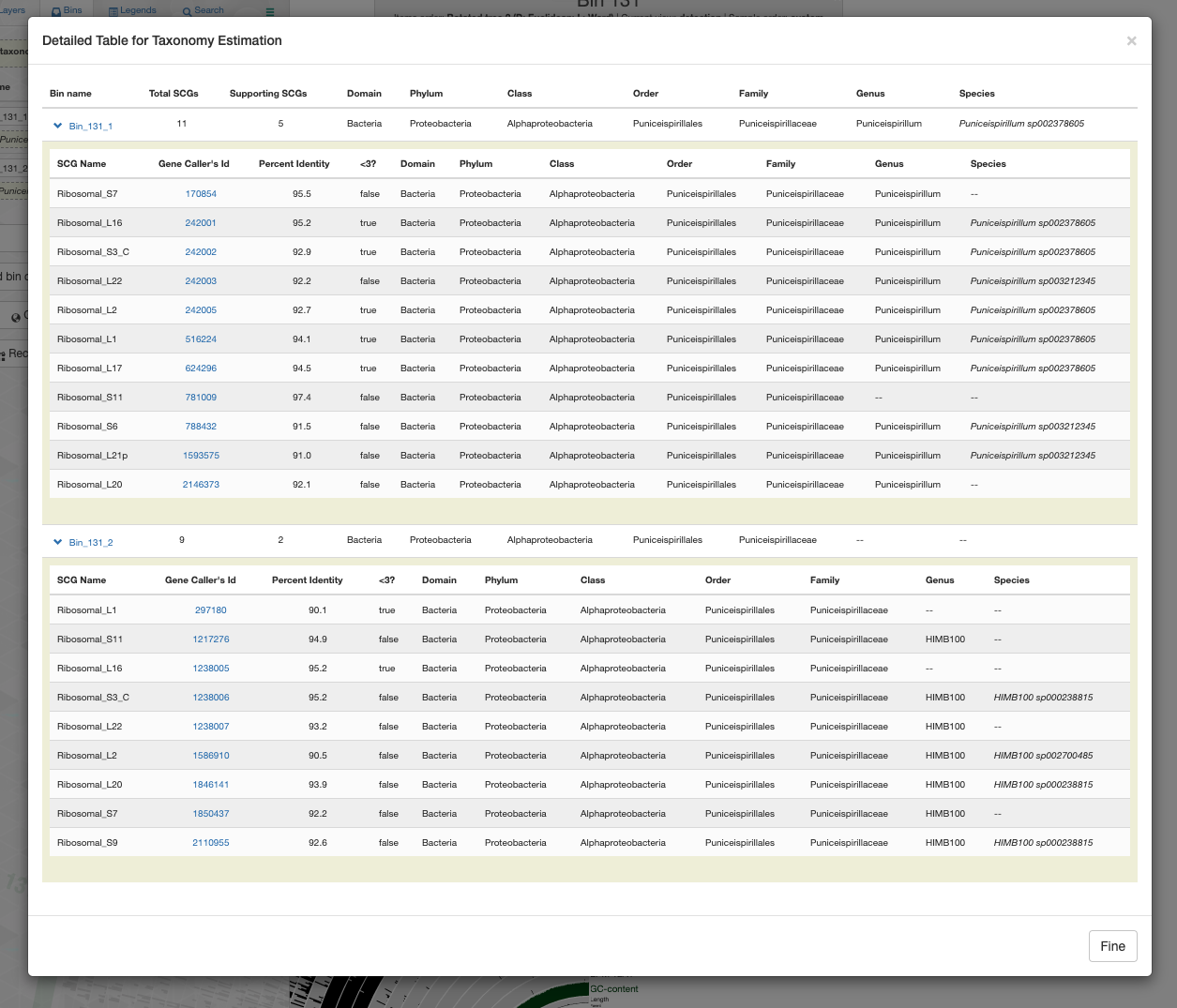
Anvi’o real-time taxonomy estimation using the GTDB resolves one bin to the genus Puniceispirillum and the other to HIMB100:
Example 5
It is not only CONCOCT that produces bins that are difficult to resolve. Here is our final example from MetaBAT:
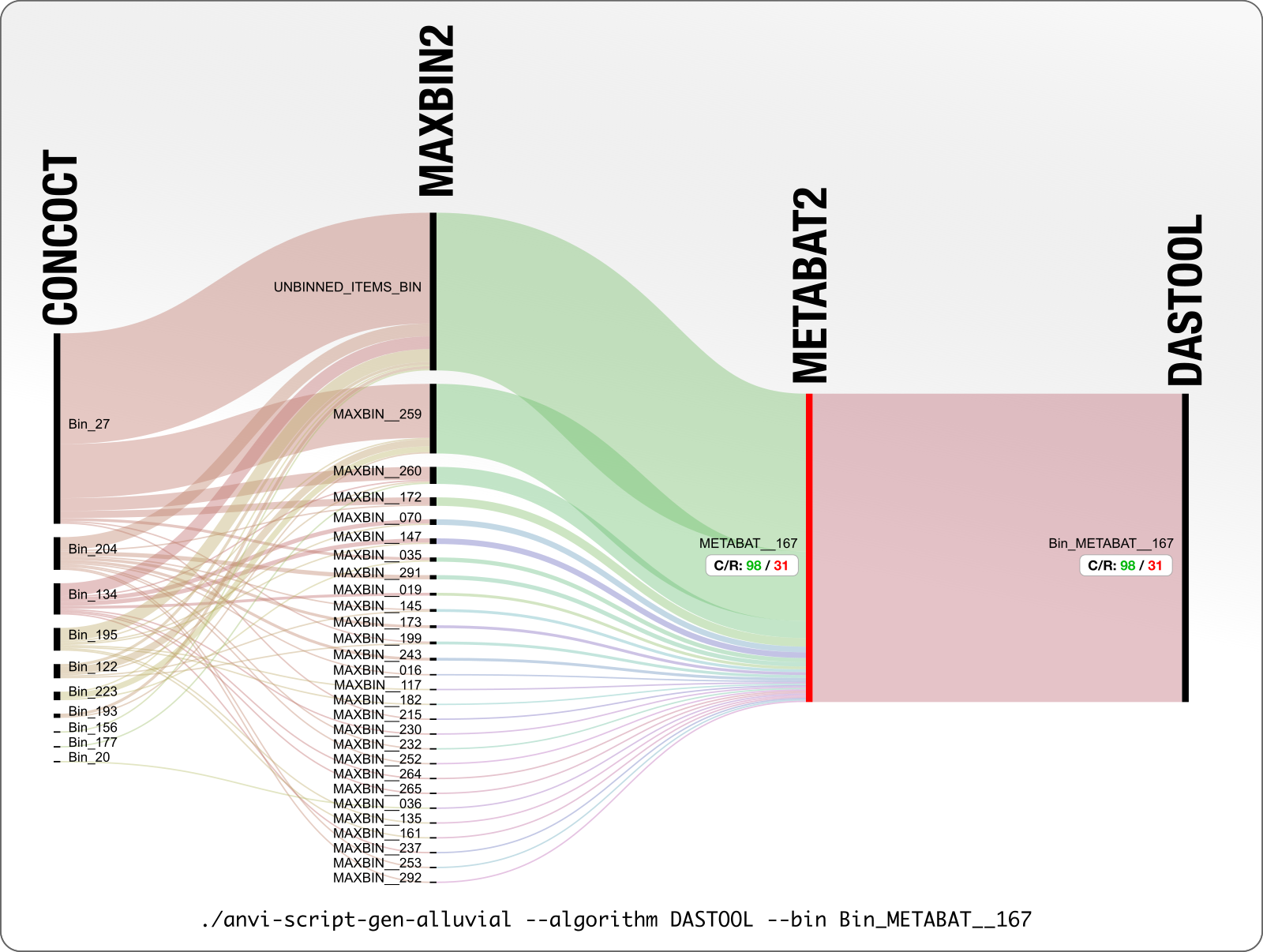
DAS Tool determines that 98% complete and 31% redundant MetaBAT bin is the best we can get from this mess. Looking at what is going on with CONCOCT and MaxBin, we wholeheartedly agree!
But we proceed to ask the same question to push for manual refinement anyway: what if we manually refined the MetaBAT bin METABAT__167 and add those results onto this display?
Here it is:
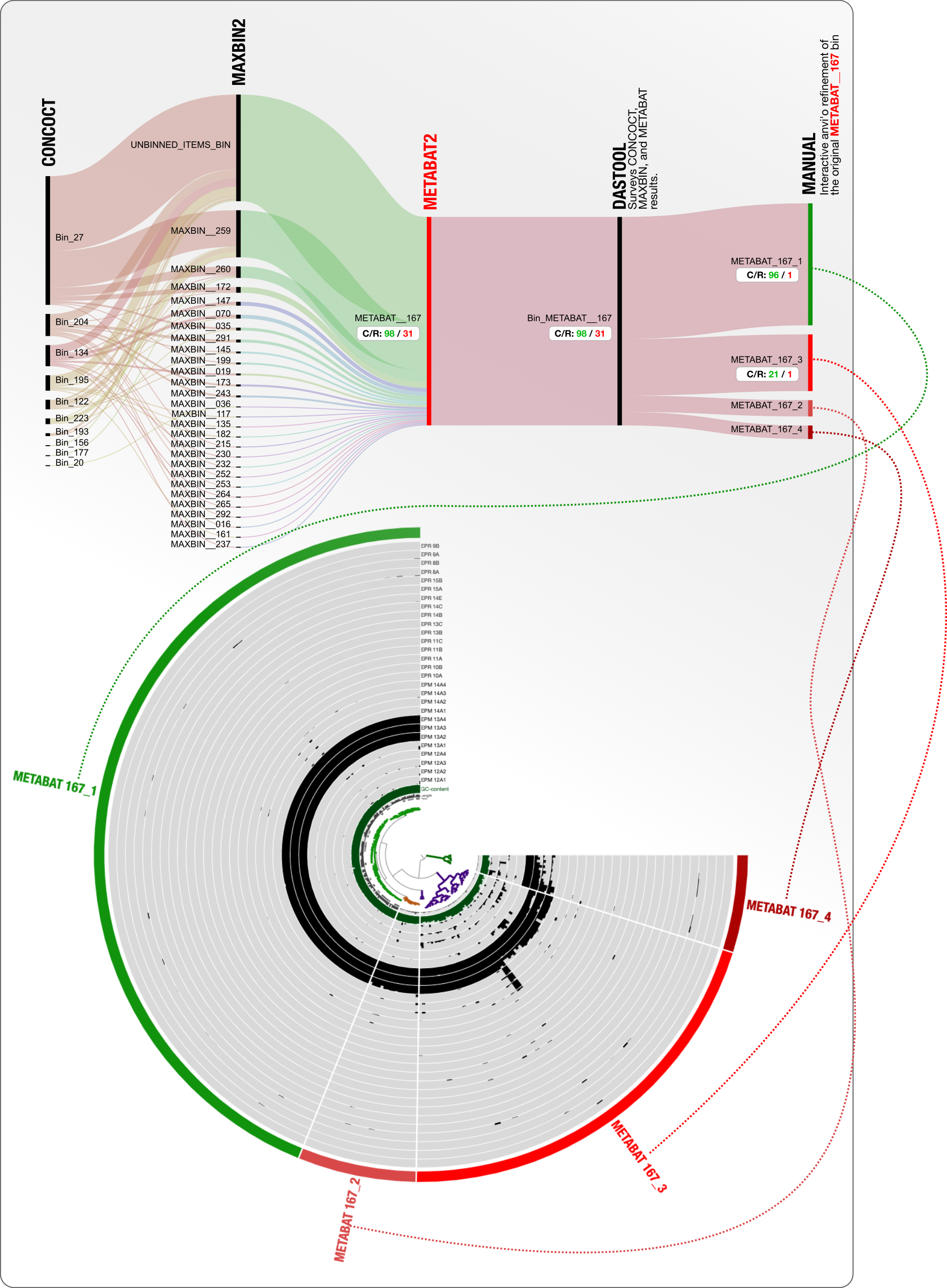
31% redundancy goes down to 1% redundancy, and from that chaos emerges an OK Erythrobacter genome to continue to stay in this analysis.
Final two cents
For those who develop binning algorithms
Please consider doing what we did here: show how your algorithm compares to others beyond summary statistics over MAG sizes or completion estimates, and through elucidating the sources of agreements, reasons for disagreements. Show what is going on inside the bins your algorithm identifies by visualizing the coverage and detection of all contigs.
Don’t trust ‘mean coverage’ blindly, because mean coverage can mean a lot of things: take a look.
This is important for two reasons. One, it is a way for users of your algorithm to know in what situations they should use it. Two, it is a way for you to see how to improve your strategy while still developing it, because seeing is everything.
For those who generate MAGs or rely on them
We can continue to use single-copy core genes to remove seemingly bad bins when they can actually be saved, and this would be wasteful.
We can assume that if we only keep the bins with good completion and redundancy estimates based on single-copy core genes the remaining bins would be clean, and this would be naive.
The last example in this post was not the final example in this dataset: visualizations in this post represent every single bin we randomly chose and looked at. They are not unique examples, they are not unrealistic.
If you are working with a relatively complex environment with a relatively small number of samples, if you are using Illumina sequencing, a popular assembly software, and a popular binning algorithm, everything you see here is more than likely happening in your dataset as well. If you are looking at binning efforts from individual samples, then you have much more reasons to be concerned.
This is true for recent publications that described large number of genomes, and these monumental efforts indeed come with their own shortcomings. For instance, take this TM7 MAG that appeared in a recent publication by Pasolli et al.:
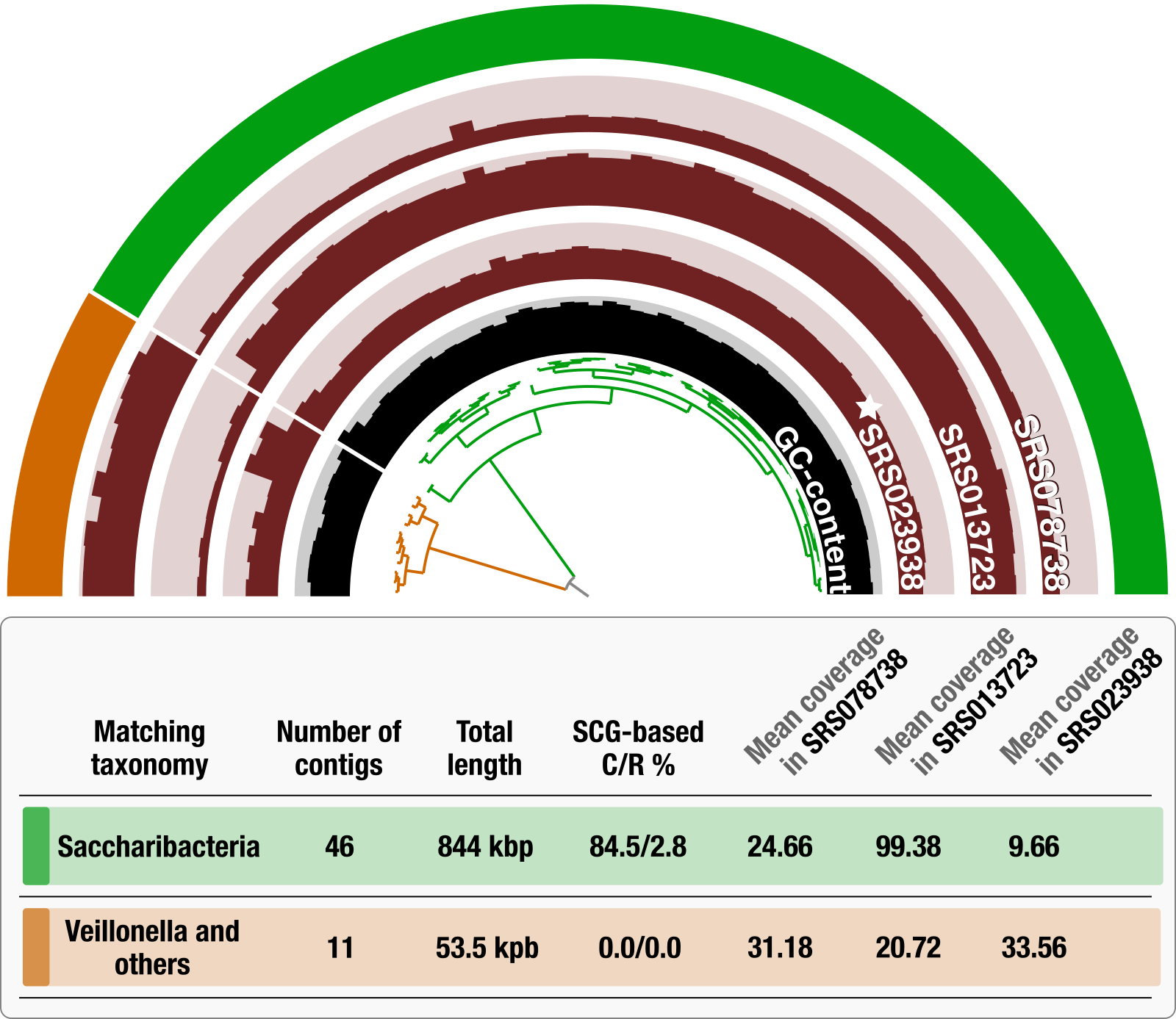
The figure is from Lin-Xing Chen et al., and the half-circle at the top displays the mean coverage of each contig in this Pasolli MAG across three plaque metagenomes that belong to the same individual, where the star symbol denotes the sample from which the original MAG was reconstructed. The dendrogram in the center represents the hierarchical clustering of the 57 contigs based on their sequence composition and differential mean coverage across the three metagenomes, while the innermost circle displays the GC-content for each contig. The outermost circle marks two clusters: one with 46 contigs (green) and another one with 11 contigs (orange). The table underneath this display summarizes various statistics about these two clusters, including the best matching taxonomy, total length, completion and redundancy (C/R) estimations based on SCGs, and the average mean coverage of each cluster across metagenomes.
Almost 20% of the contigs in this MAG is contamination from Veillonella and other oral taxa. These contigs ended up together, because in the only plaque sample that was used for binning (SRS023938), their coverages were relatively comparable.
The reason to highlight this bin is not to criticize Passoli et al.’s work. There was nothing much anyone could do for these 11 contigs when there were millions of others spreading through 150,000 genome bins to deal with. But the reason is to show that this is the reality of large-scale fully-automated binning efforts in the midst of all the shortcomings of metagenomics.
Overall, it is essential to be aware, but there is no need to be pessimistic.
Science moves on despite all these difficulties, and moves on fast. We are thankful for the passion and dedication of those who develop binning algorithms, create new public metagenomes, and put their best effort to catalogue the genomic make up of natural habitats, such as Passoli et al.
And thank you for reading all the way down here.
A new blog post from Jarrod Scott and I in which we try to take a look at what is going on in some MAGs by visualizing the contig membership changes across binning strategies that were applied to the same data:https://t.co/u92wz8QSTt
— A. Murat Eren (Meren) (@merenbey) January 2, 2020
Happy new year!