
Table of Contents
- Completion and redundancy
- > 90% completion + < 10% redundancy ~= Golden
- A short story from 4,000+ complete bacterial genomes
- Identifying contamination is one thing, dealing with it is another
- F.A.Q. (each answer is worth 2 cents or less)
- I have a bin with more than 10% redundancy. What do I do?
- My bin was C63%/R16%, I refined it and now I have two bins: C44%/R4% and C40%/R7% :(
- Anvi’o soft-splits contigs, does it count BSCGs twice?
- Can anvi’o tell me which splits contain duplicate BSCGs?
- When I remove one split, redundancy goes from 18% to 7%! :)
- I have a question that is not here :/
Completion and redundancy
Every time you select a bunch of contigs into a genome bin through the interactive interface, anvi’o tells you in real time about the estimated level of completion and redundancy of your bin. To do that, anvi’o uses four previously published bacterial single-copy core gene (BSGC) collections. Although there are currently four BSCGs in the repository, we have begun to be very fond of the collection by Campbell et al., which contains 139 BSCGs.
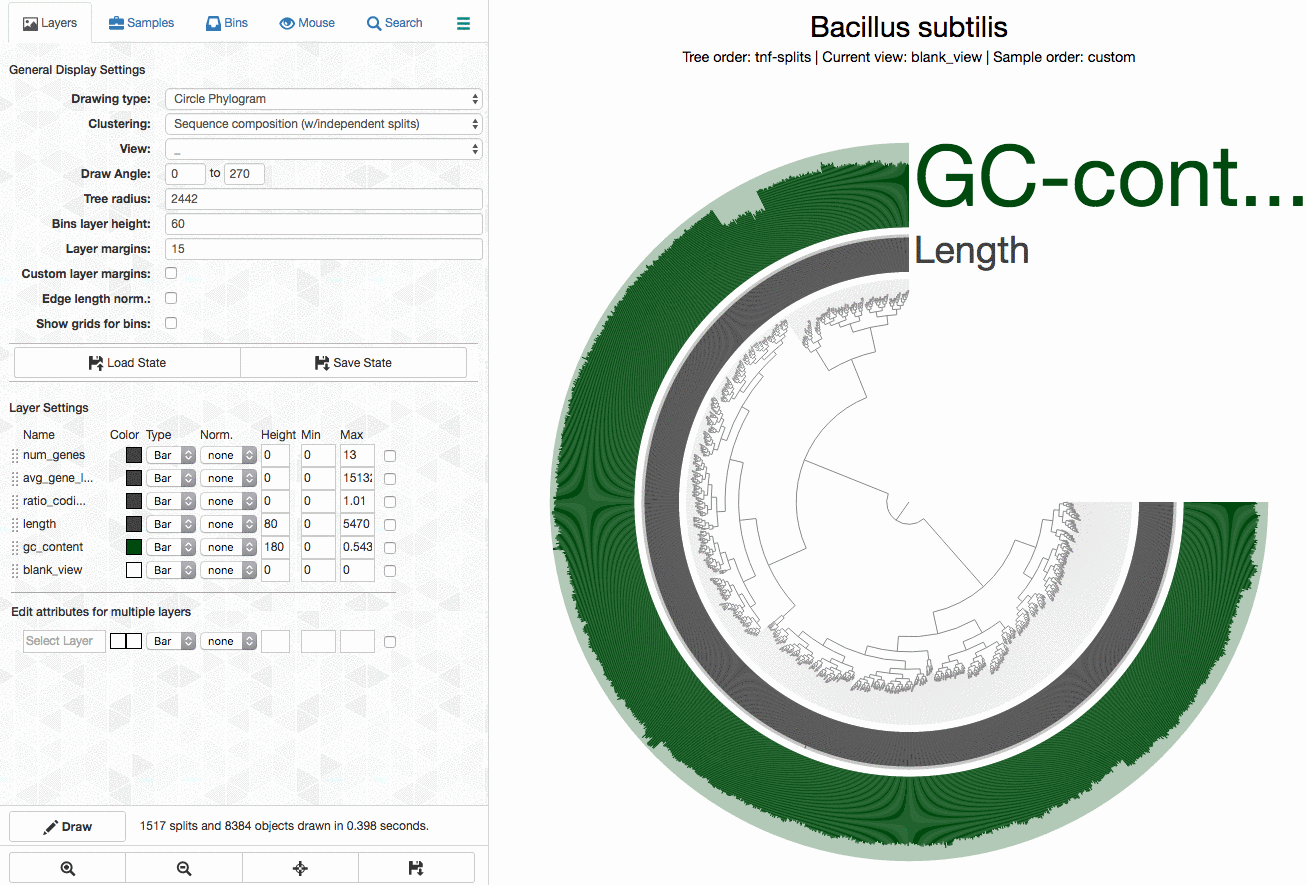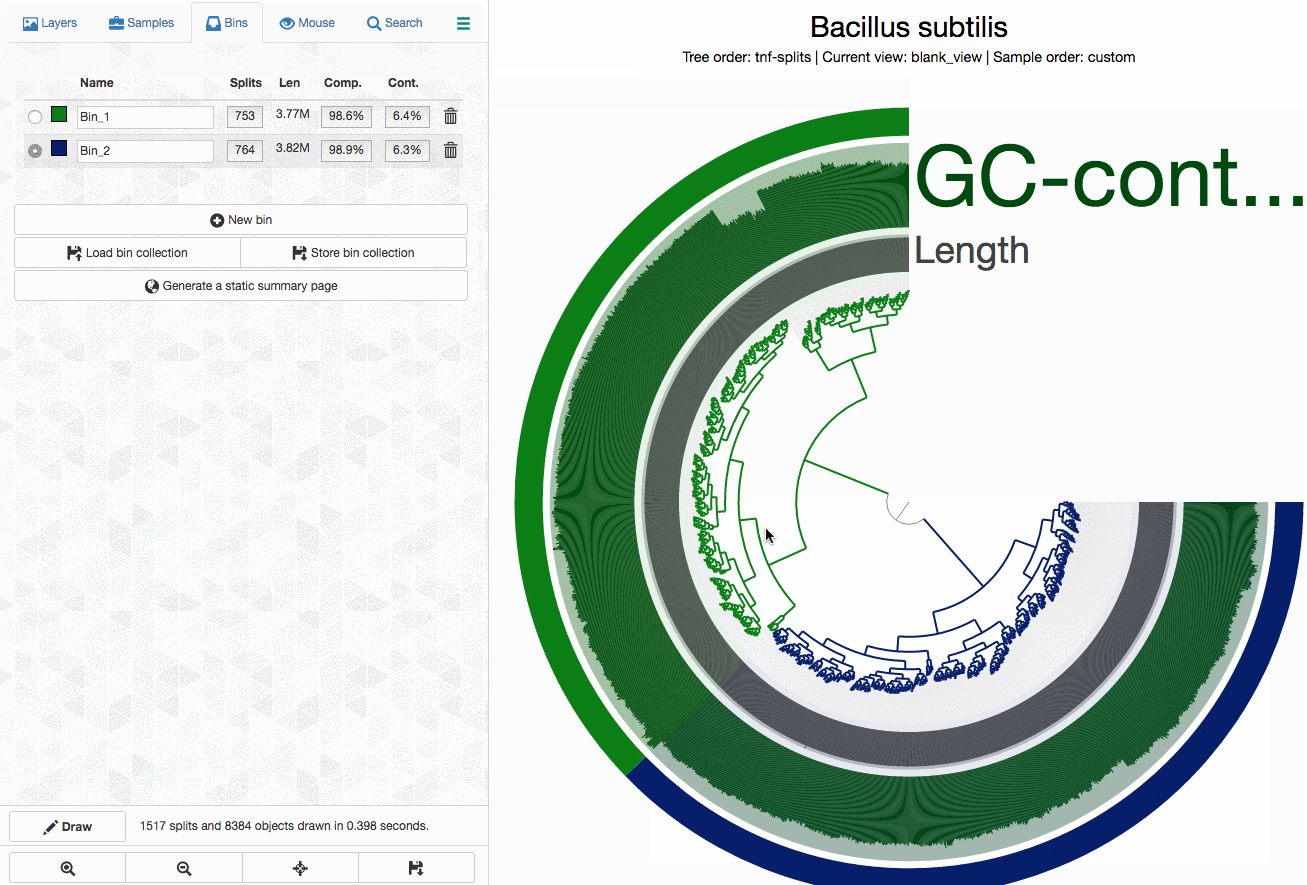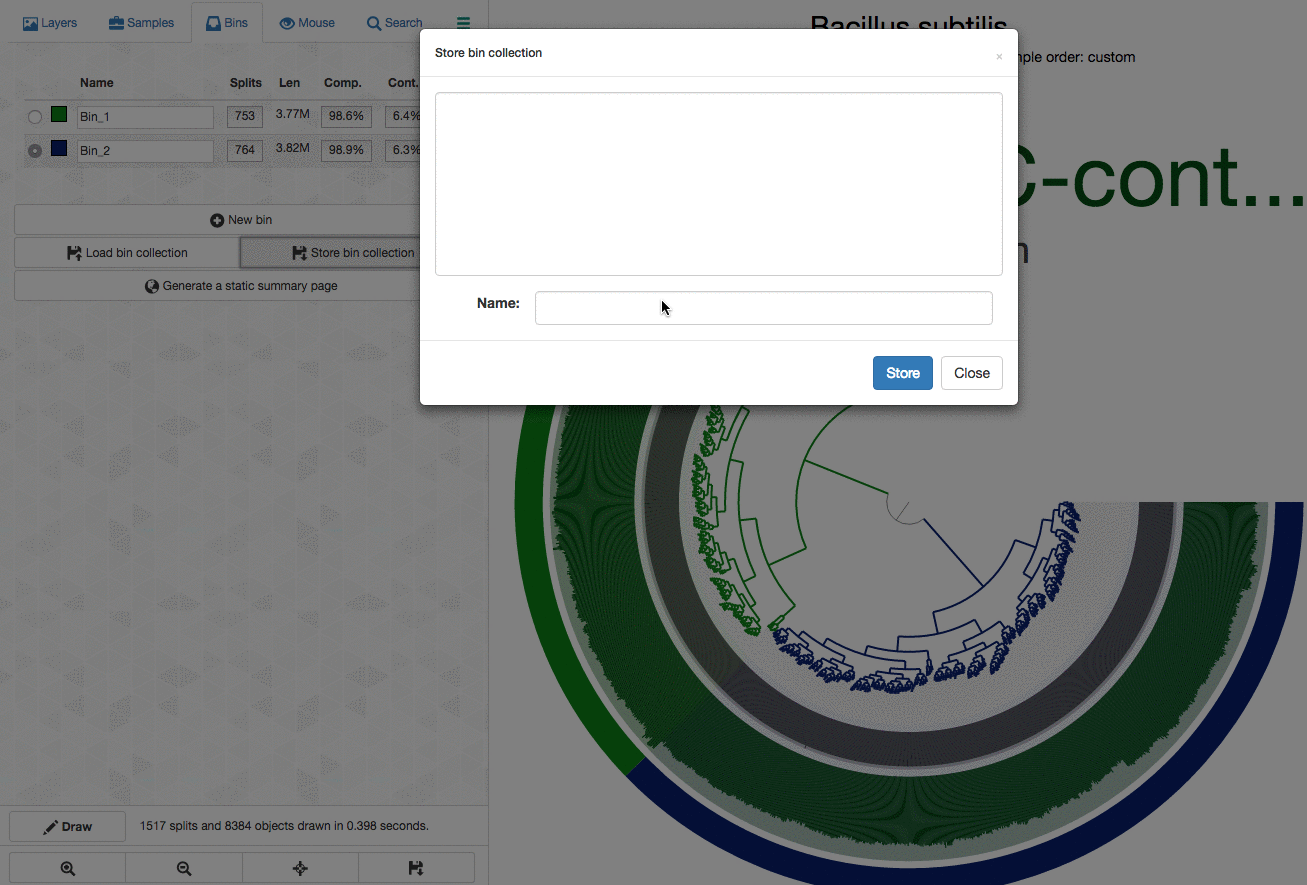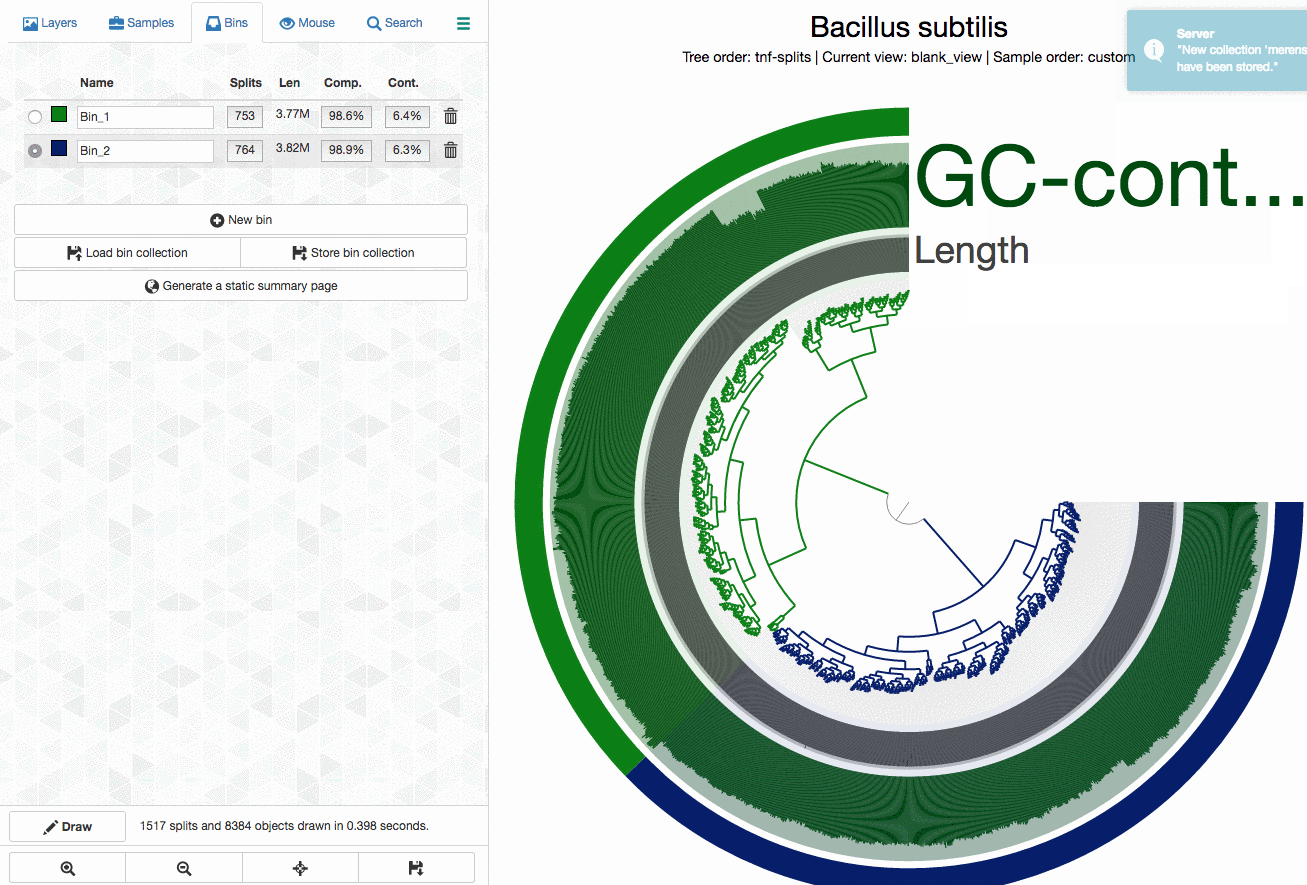{kind=link}
Why do we call it redundancy and not contamination? So there are a number of genes that occur once in most bacterial genomes that we managed to isolate. If we find multiple copies of those genes in a bin, we start to feel a bit uneasy. The term ‘redundancy’ and the term ‘contamination’ are inherently linked, but they are quite different: the level of redundancy of BSCGs in a genome bin is a result of an objective observation, contamination, on the other hand, is a suggestion we make (which, in most cases, whether the accusation has real basis or not goes untested). If a bacterial genome that did not end up in our cultivars has multiple copies of one of those many so called single copy genes, it wouldn’t mean it is ‘contaminated’. Therefore we avoid using the term ‘contamination’ until there is more evidence –and this post is partially about identifying the cutoff for that evidence. Some history to make sure the credit goes to where it should: In the early days of anvi’o we were using ‘contamination’ everywhere in the codebase and user interfaces. Then we ran into Jed Fuhrman one day, and he straightened us up. So it went.
Although we find it extremely helpful to have real-time estimates available to us while working with our metagenomic bins, it usually is a struggle to make sure these bins are coherent, i.e., they have as many BSCGs as necessary, but not more. However, one thing you quickly learn when you start working with metagenomes is that nothing ever is ideal in these complex data, and Murphy here is not a wise old guy, but a supervillan who really sticks with his evil-er motto.
Anything that can go wrong, will go wrong, and then some other perfectly OK stuff will go wrong, too. And there is nothing much you can do about it either. Yes. Yikes.
When it is clear that pretty much everything can go wrong, it becomes even more critical to be scrupulous with the binning step. Because binning is the first step in the metagenomic workflow after sampling where everything is (or can be) under your control, and it is the step where you can have a beautiful collection of genomes for your downstream analyses.
Due to numerous issues related to the assembly process, metagenome-assembled genomes (MAGs) are rarely 100% “complete” with respect to the occurrence of BSCGs out-of-the-box. Due to some additional issues related to mapping and automatic binning steps, as well as occasional non-specific hits to shorter BSCGs, our MAGs rarely have only one copy of every BSCG. So 100% completion and 0% redundancy with respect to BSCGs is not realistic. But then, what is realistic? When is it OK to stop refining a bin, and when is it not OK to say “well, this is the genome bin I will call final, analyze, and publish”?
> 90% completion + < 10% redundancy ~= Golden
We usually suggest that in order to report a MAG it should meet both of these minimal criteria:
- More than 50% complete, or more than 2Mb in size1.
- Less than 10% redundant based on Campbell et al.’s BSCGs2.
1 Our observations indicate that the presence of closely related organisms cause dramatic underestimations of completion, probably because contigs tend to break up around relatively more conserved BSCGs across multiple genomes. 2 This clearly will only work properly for bacterial genomes –you should check this post by Mike Lee for an archaeal collection.
Although both points are important, I think the second point is a bit more important: making sure MAGs are not more than 10% redundant. It would be sad to over-split an otherwise fine MAG, but it is detrimental if a MAG describes a collection of contigs that clearly originate from different genomes. And if your bacterial MAG has more than 10% redundancy, you can be almost certain that your bin is a crappy one and it contains contigs that originate from more than one genome. You can either refine it (which can give you a more concrete understanding of what might be going on in that bin), or discard it, but you really should not contaminate public databases with it.
A short story from 4,000+ complete bacterial genomes
Here is some justification for these numbers, especially for the ‘max 10% redundancy’ suggestion.
To see how well anvi’o is doing with Campbell et al’s collection, I downloaded 4,022 closed genomes from the NCBI. Then I removed one Bacillus subtilis genome from this collection, because it was 100%+ redundant. In fact I mentioned this particular and rather lovely genome in this post, but you shouldn’t spend more time on this because you have so many other things to do.
I analyzed the rest of the 4,021 genomes the same way I described here, and recovered BSCG hits for each one of them.
The following figure shows the completion, redundancy, genome size, and the number of genomes included in this analysis per phylum:
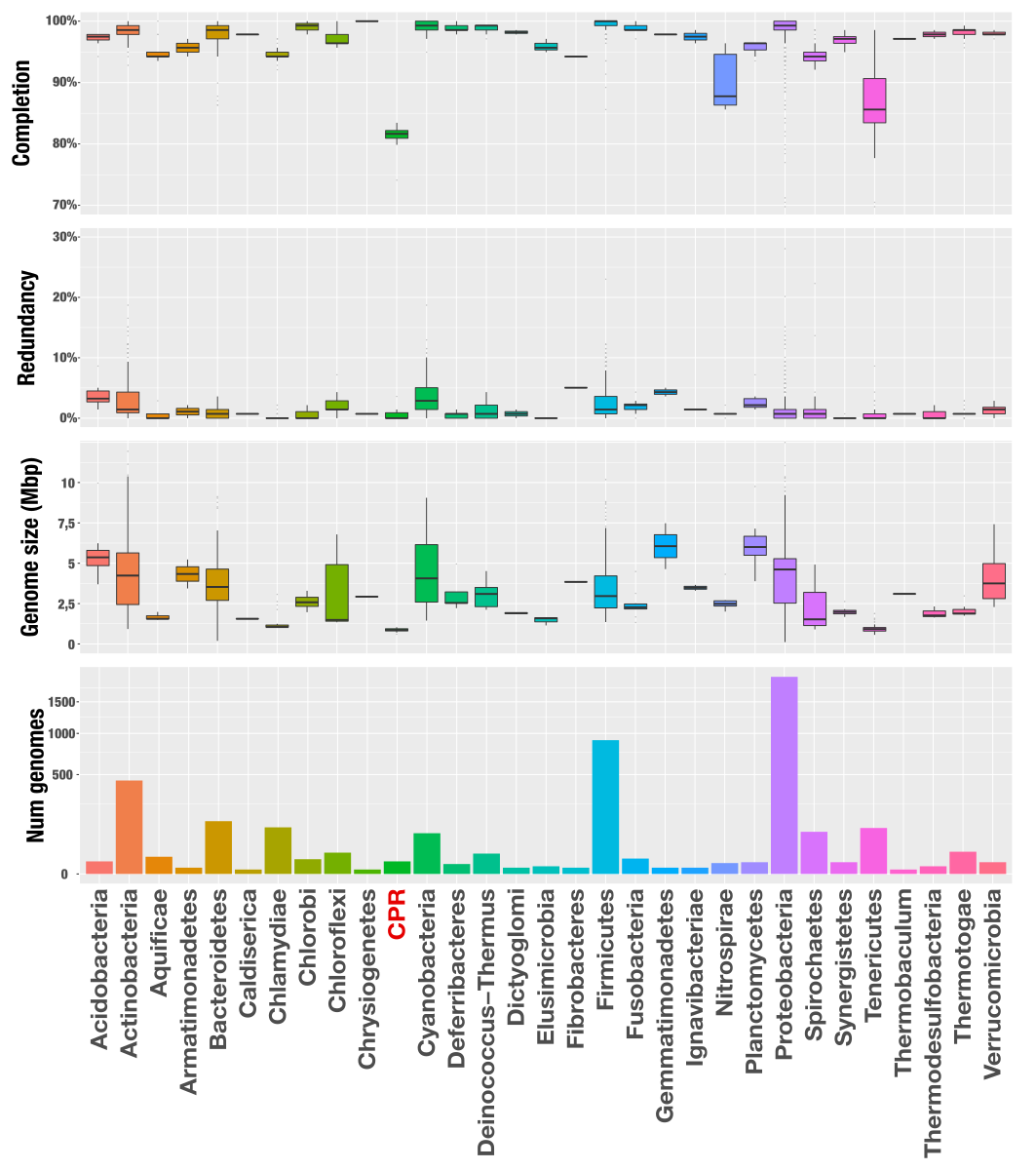
As you can see, there is a great drop in completion estimates for Candidate Phyla Radiation (CPR) genomes. Which is not surprising. I had already analyzed them with the same 139 BSCGs in the post “Predicting CPR genomes”, and was expecting to see that. Apart from CPR’s, the phylum Nitrospira, and Tenericutes have lower completion estimates based on Campbell et al.’s BSCGs. There is a clear taxonomical signal, and some complete genomes that belong to certain taxa miss more BSCGs compared to others. Not very surprising, and not very concerning either. Just a good reminder that lack of completion does not always mean the genome is not quite well put together, since all BSCGs are originating from isolates, and we know all the biases associated with cultivation.
On the other hand, regardless of genome size or completion estimates, redundancy estimates remain below 10% in general. In fact the vast majority of the genomes have less than 5% redundancy. I will later look into these much more carefully to maybe suggest some updates to the collection. But for now, here is a better way to represent what is going on for everything:
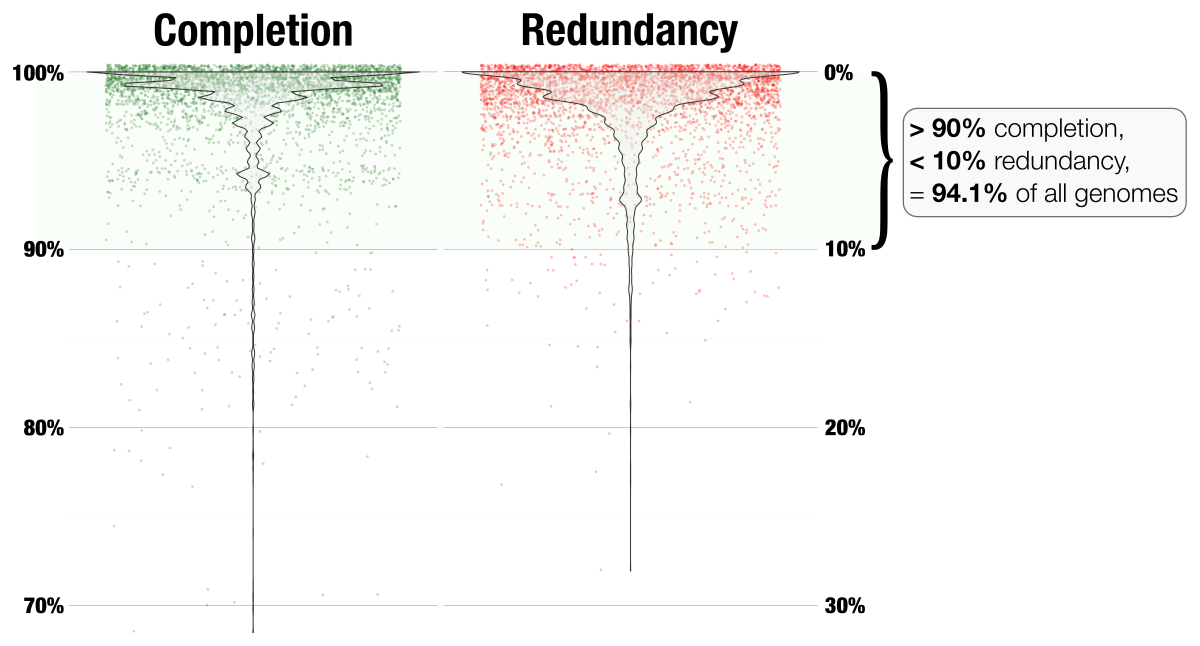
Every dot in this figure represents one of the 4,022 genomes in this analysis. So 94% of all closed genomes in the NCBI has redundancy way less than 10% based on this BSCG collection.
A lower completion score may be due to underestimation, but a higher redundancy will almost never be due to overestimation. You must try to clean it up.
This can be your golden rule, and maximum 10% redundancy would be a way to make sure you are not shooting yourself in the foot.
Identifying contamination is one thing, dealing with it is another
One of the groups we learn a lot from is the one that is led by Gene Tyson at the Australian Centre for Ecogenomics. One of the tools that came from Tyson’s group, CheckM, is a powerful solution to assess the completion of genome bins and the redundancy trapped in them. Besides the tool, the methods paper for CheckM (Parks DH et al., 2014) provides a lot of insights, and you should read it, if you haven’t, to have a more complete perspective on how to make sure our MAGs are appropriate.
One can identify contamination in multiple ways. However, dealing with it is a different story. It requires interaction and care. Make no mistake: you will get garbage from metagenomic assemblies. But there are ways to make sure you have the best possible MAG collection from your study to report. If you want to see how far you can go being stringent with your MAGs, I suggest you take a look at publications coming from Jill Banfield’s group.
Anvi’o has been very helpful for us to better understand what is happening in our MAGs, and allowed us to report coherent genome bins. Here are three other blog posts in which we demonstrated how to do these things with anvi’o:
- Refining a bin using anvi’o
- Removing contaminants from cultivars with anvi’o
- Binning without mapping
F.A.Q. (each answer is worth 2 cents or less)
I have a bin with more than 10% redundancy. What do I do?
Either refine it, or remove it.
My bin was C63%/R16%, I refined it and now I have two bins: C44%/R4% and C40%/R7% :(
Sad. But don’t be upset. It seems it was a garbage bin, and now you are free.
Anvi’o soft-splits contigs, does it count BSCGs twice?
No. Anvi’o v1 branch follows unique gene IDs to make sure splits do not cause inflation of observed BSCGs. Anvi’o v2 branch is even more stringent: it does not allow you to cut genes in half (unless you revolt against it’s authority and use --skip-mindful-splitting flag while generating your anvi’o contigs database).
Can anvi’o tell me which splits contain duplicate BSCGs?
Yes, it can. You can interactively see which splits in a bin contains multiple hits. Click on the image for an animation:
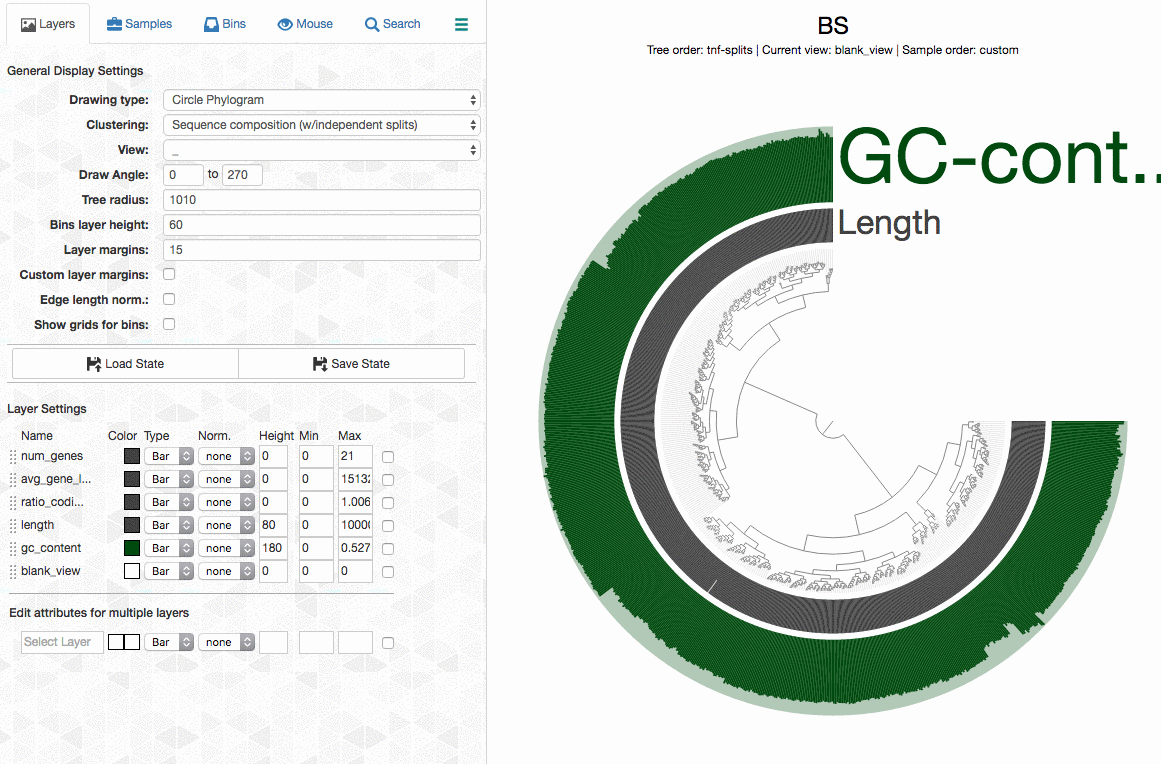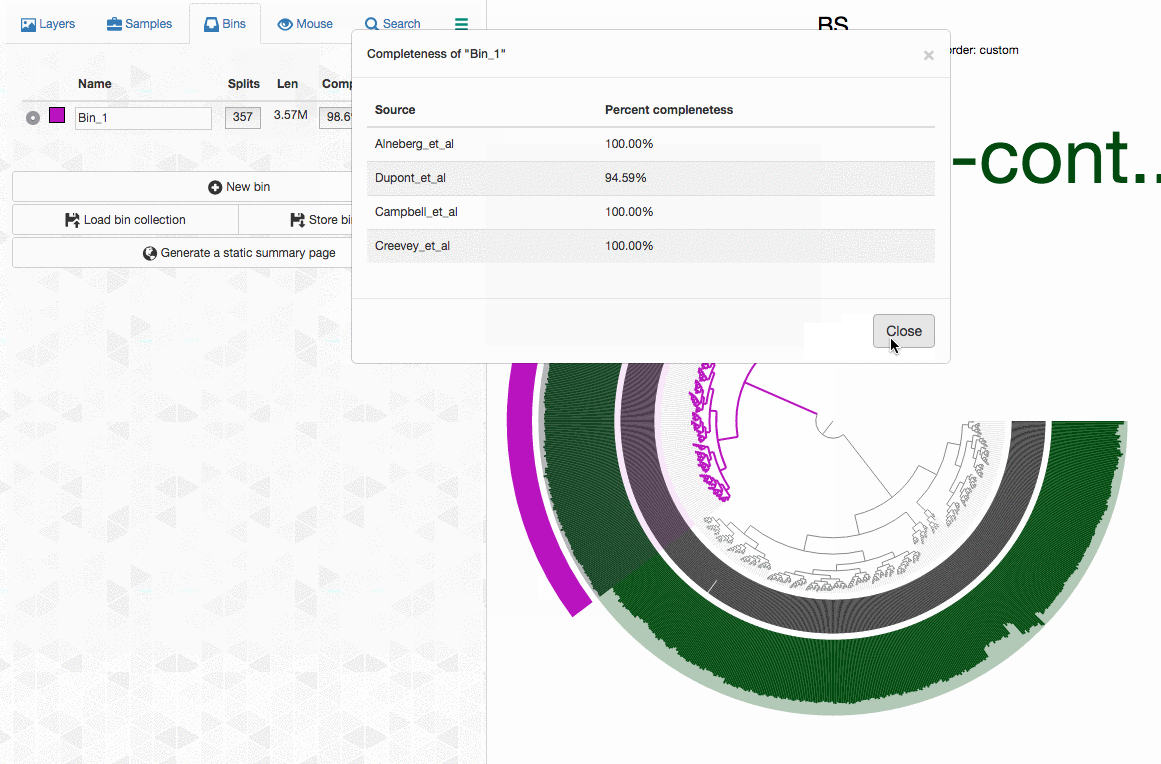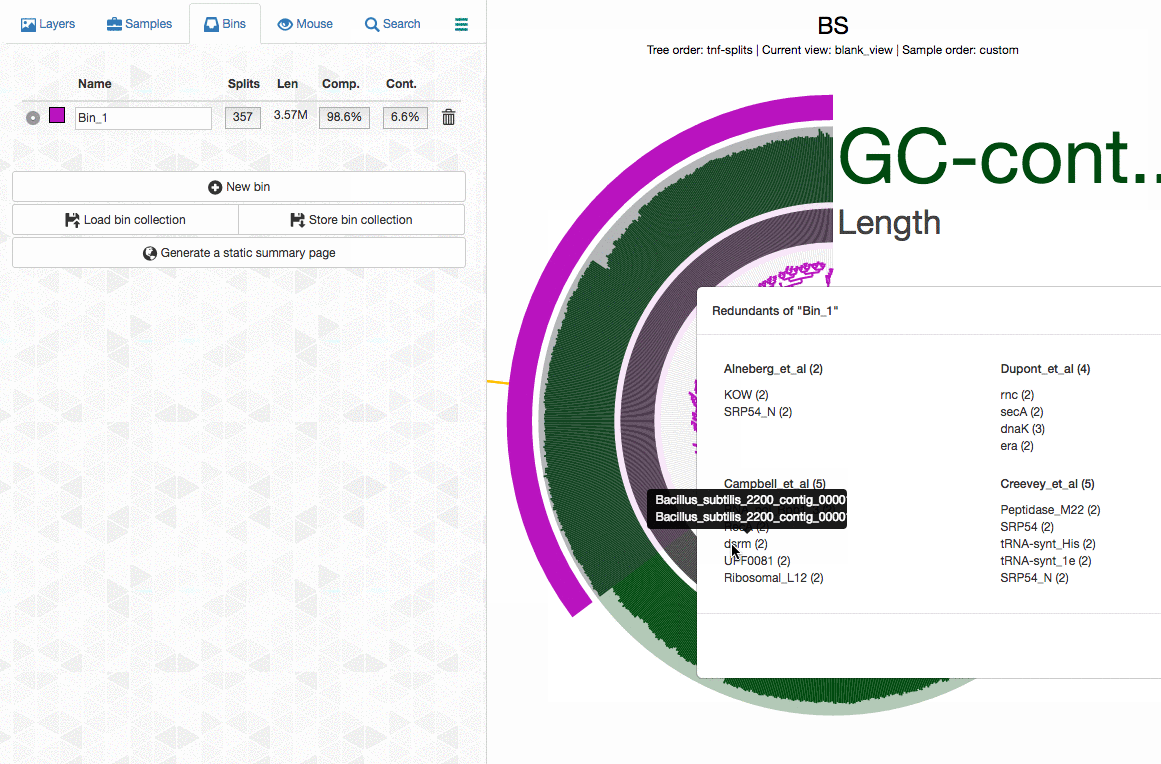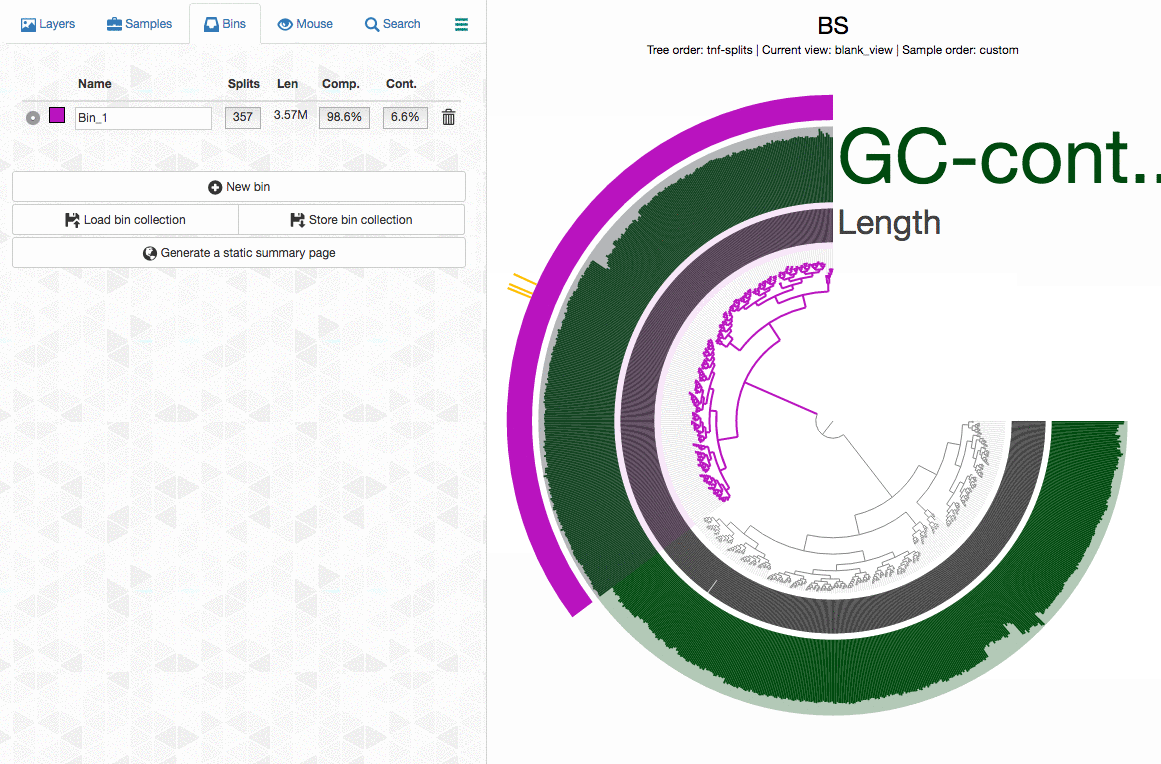
When I remove one split, redundancy goes from 18% to 7%! :)
It is very critical that you don’t play this game. BSCGs should not be the driver of how you identify your bins, or refine them. For this purpose, you must solely rely on differential coverage and/or tetra-nucleotide frequency. Otherwise you can hand-pick every split to perfect completion / redundancy estimates, which would look great on the paper without any biological gain. Please consider this: the purpose is not to get those estimates to a perfection. The purpose is to use them to make sure the binning is not glaringly inapproripate.
I have a question that is not here :/
Great! Please write it down in the comments section, or send us an e-mail, and we will see what we can do!
BSCGs are great tools to shed light on the plethora of unknown, they should be used wisely. They shouldn’t be given too much credit when they suggest lower completion estimates because they may be wrong (see CPR genomes). But they should’t be ignored when they suggest high redundancy, because 95% of the time they will be correct.
All these aside, we recently managed to address one of the most sorely missed need of every software platfrom for anvi’o:
We heard that #anvio will not be taken seriously unless it has a sticker. Well, we got that covered: pic.twitter.com/24IVCDQqrC
— A. Murat Eren (@merenbey) June 6, 2016
Just so you know.