
Table of Contents
Meren’s note on 2020: If you are here to learn about how to create a user-defined HMM source for anvi’o, please find the most up-to-date documentation for it here.
Meren’s note on September 2016: Starting from v2.0.3, the archaeal single-copy core core gene collection released by Rinke et al., and described in this post by Mike Lee, is included in anvi’o. Details of this transition can be viewed here. We thank Mike for helping us to improve anvi’o.
Meren’s note on May 2016 (updated Dec 2018): The following post is kindly contributed by Mike Lee, who is a microbial ecologist/bioinformatician and currently a postdoc with NASA at the Ames Research Center. His research spans from basalts at the bottoms of our oceans up to the International Space Station, and he also runs a website designed to help biologists get into bioinformatics. Mike’s post made me realize that there could be some improvements in the codebase to make better use of the archaeal single-copy gene collection he used for his project, and I promise to improve the anvi’o codebase to be less biased towards bacteria in the future ;) I thank Mike for sharing his experience with everyone through this post, and for his very kind words about anvi’o.
One of the major strengths of anvi’o is the capability of manually curating your genome bins with real-time updates of percent completeness and contamination estimates. The information necessary to estimate completeness comes from the scanning of your contigs using previously published bacterial single-copy gene collections. When you run the following command following the standard metagenomic workflow, the occurrence of bacterial single-copy genes across your contigs is all added to your contigs database:
$ anvi-run-hmms -c contigs.db
As of today, when anvi-run-hmms is run without providing any further arguments, it automatically utilizes the four HMM profiles included in anvi’o codebase. These profiles are quite useful and convenient for bacterial genomes. However, if you are purposely trying to refine archaeal bins, or perhaps if you are working with a lab that has spent some time and effort to build their own single-copy gene collections, the ability to change the underlying HMM single-copy gene collections would certainly help. Fortunately, anvi’o has an easy way to specify which collection(s) to consider when working in the interactive mode.
As noted above, running the anvi-run-hmms program with no additional arguments other than your contigs database will scan for the four preloaded bacterial single-copy gene collections. If you would like to use another collection, you need only to add the location of the directory that contains your HMM profiles:
$ anvi-run-hmms -c contigs.db -H Rinke_archaeal_HMM/
Anvi’o will expect the directory denoted by the -H flag above to contain six special files:
| File Name | Content and Purpose |
|---|---|
| genes.hmm.gz | A gzip of concatenated HMM profiles. |
| genes.txt | A TAB-delimited file containing three columns; gene name, accession number, and source of HMM profiles listed in genes.hmm.gz. |
| kind.txt | A flat text file which contains a single word identifying what type of profile the directory contains. If this word is ‘singlecopy’, the profile is used to calculate percent completeness and contamination. Otherwise it will only be used to visualize contigs with HMM hits without being utilized to estimate completeness. |
| reference.txt | A file containing source information for this profile to cite it properly. |
| target.txt | A file containing the target alphabet and context. See this for more details. For this particular collection the target will be AA:GENE (because it was prepared using amino acid alignments, and we want them to be searched within gene calls stored in contigs databases), however, the the target term could be any combination of AA, DNA, or RNA for the alphabet part, and GENE or CONTIG for the context part (well, except AA:CONTIG, because we can’t translate contigs). |
| noise_cutoff_terms.txt | A file to specify how to deal with noise. See this comment for more information on the contents of this file. |
Examples of each file can be found here, and if you’d like to jump right in using the archaeal single-copy gene collection by Rinke et al., please help yourself to the directory located here.
The following figure compares the completeness and contamination estimates for 5 archaeal and 3 bacterial genomes I pulled from IMG. For each genome, the figure displays the estimations based on the default bacterial single-copy gene collections distributed with anvi’o, and Rinke et al.’s archaeal gene collection I added to my contigs database:
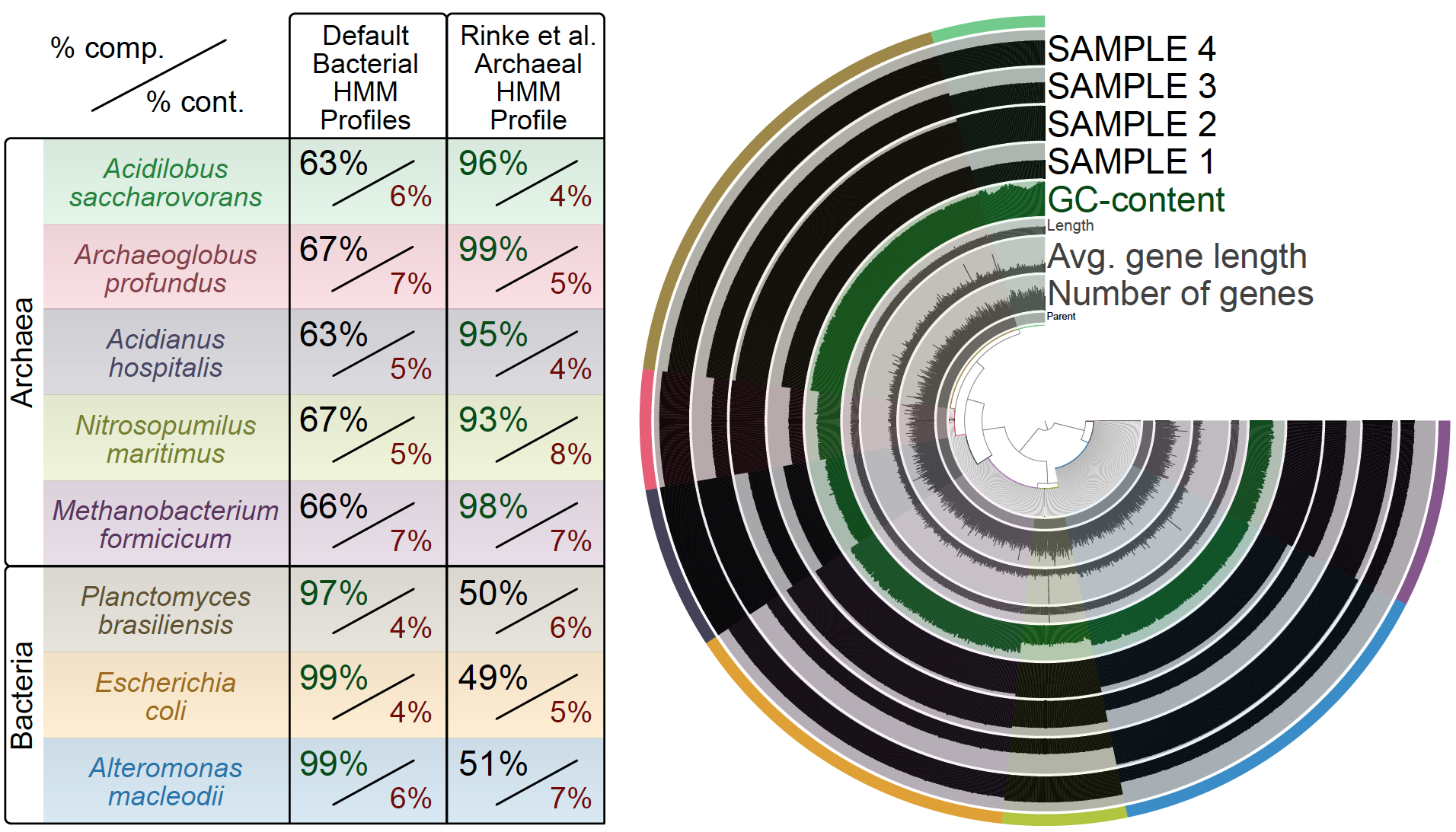
Not all that surprisingly, percent completeness estimates for the archaeal bins are rather poor when they are based on HMM profiles for bacterial single-copy genes. In contrast, when the archaeal profile is used, they are much more accurately evaluated.
If you are anything like me and many other fortunate researcher out there, you have already realized that not only is anvi’o incredibly user-friendly, which of course is nice, but also it can be an extremely powerful tool. This is largely due to it being designed with an underlying framework that can be easily manipulated (in small part exemplified here) and built upon. To be sure, while most of us are still barely scratching the surface as far as leveraging anvi’o’s full potential, the team is busy adding more and more exceedingly wonderful functionality - definitely check out the CPR hunting and pangenomics posts if you haven’t yet!