
Table of Contents
A note from the Meren Lab: We are very thankful to Veronika for taking the time to share her experience with anvi-refine. Veronika is a graduate student at the University of California, Santa Barbara. She is affiliated with the Valentine Lab, where she studies microbial mat rings in deep ocean sediment using metagenomics.
I have been investigating ways to refine metagenome-assembled genomes (MAGs) after finding out an interesting genome bin to be a mixed bin, as a result of imperfect automated binning.
I used to think that if a draft genome had >90% completion and <10% redundancy, things were good. This isn’t necessarily true: the completion and redundancy values are one of several aspects of a bin that we can look at, and this is what my post attempts to demonstrate.
Before I start, this is what I read originally when I started refining my MAGs and I recommend you to take a look, as well. Another relevant post on single-copy core genes and HMM collections is here.
My anvi-refine notes
The screenshots from my work are included below with some notes.
I use the shorthand C/R for completion and redundancy, and for the rest of the post, I will use “C90/R10” notation to describe “90% completion and 10% redundancy” for a given bin. The idea behind C/R is explained much better elsewhere, but very basically: we can use the single-copy core genes to estimate the completion of a genome bin. Multiple occurrence of single-copy core genes would increase the redundancy estimate.
I started by running anvi-interactive in collection mode after completing the anvi’o metagenomics workflow:
anvi-interactive -p /PATH/TO/PROFILE.db -c contigs.db -C CONCOCT
Which displayed all my bins in the CONCOCT collection with their completion and redundancy estimtes:
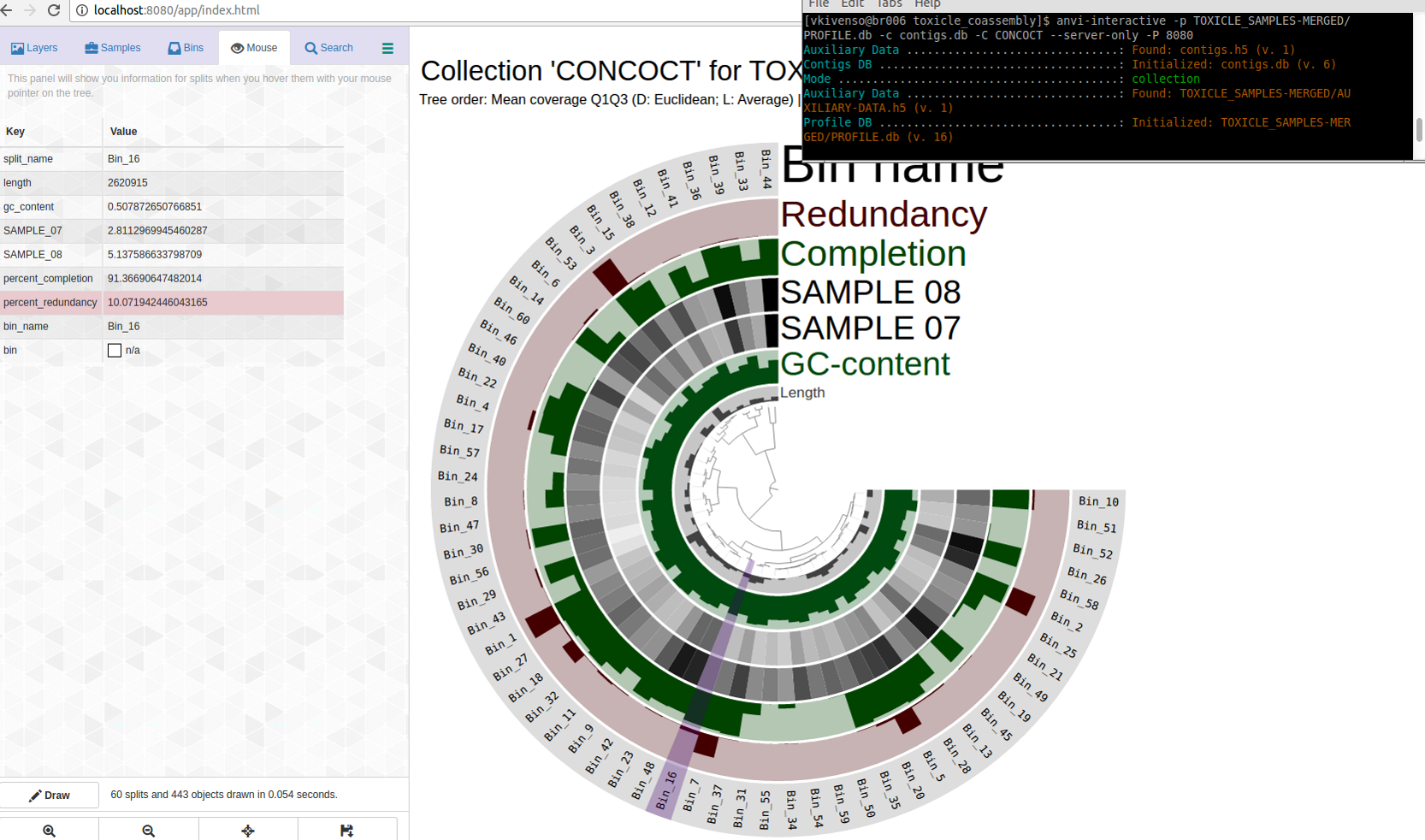
The purple highlights Bin_16.
Bin_16 is C91/R10. Not bad, maybe?
Here I learned that even bins with almost great completion and redundancy estimates may need manual refinement. Here are some examples of refinement in action.
Is Bin_16 (C91/R10) a good bin?
We switch to anvi-refine to focus on this single bin:
anvi-refine -p PATH/TO/PROFILE.db -c contigs.db -C CONCOCT -b Bin_16
Bin 16 is shown in the dendrogram below. Parts of it do not belong (circled in red), suggesting the refinement step was critical for this bin:
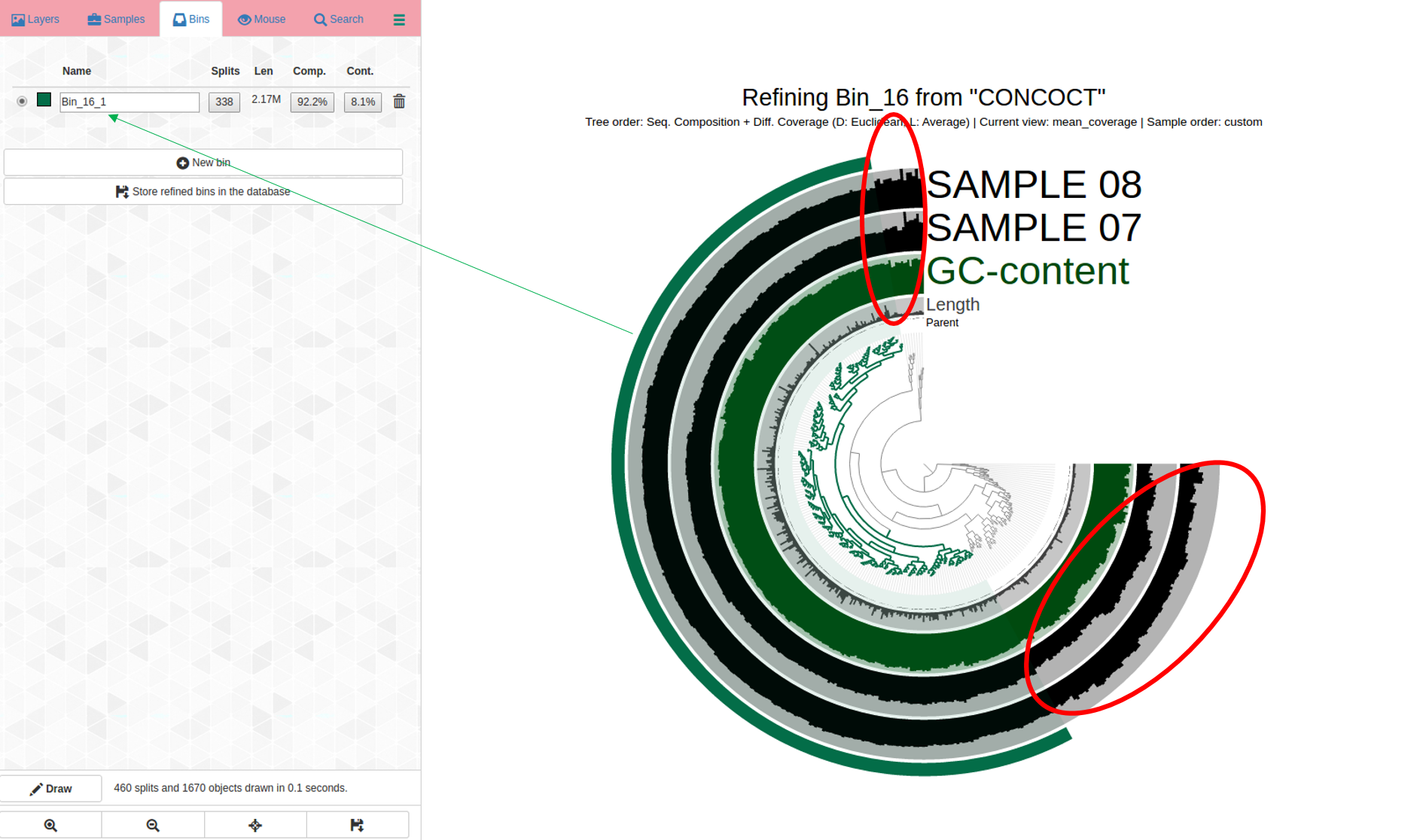
Contigs circled in red were incorrectly placed here and do not belong in this bin. We know this because the coverage (shown in the two SAMPLE 7 and SAMPLE 8 layers in black) is inconsistent; which is a sign that contigs have been incorrectly assigned to this bin, which should not include them.
The soon-to-be “new” bin, is highlighted in green + on the outermost layer; more refinement may be needed and it is worth a look to see how this first pass of refinement of the original bin looks like by running anvi-refine again.
So, more refinement, after clicking “store refined bins in database” and waiting for the interactive display to acknowledge this; anvi-refine was re-run on the new bin.
anvi-refine -p PATH/TO/PROFILE.db -c contigs.db -C CONCOCT -b Bin_16_1
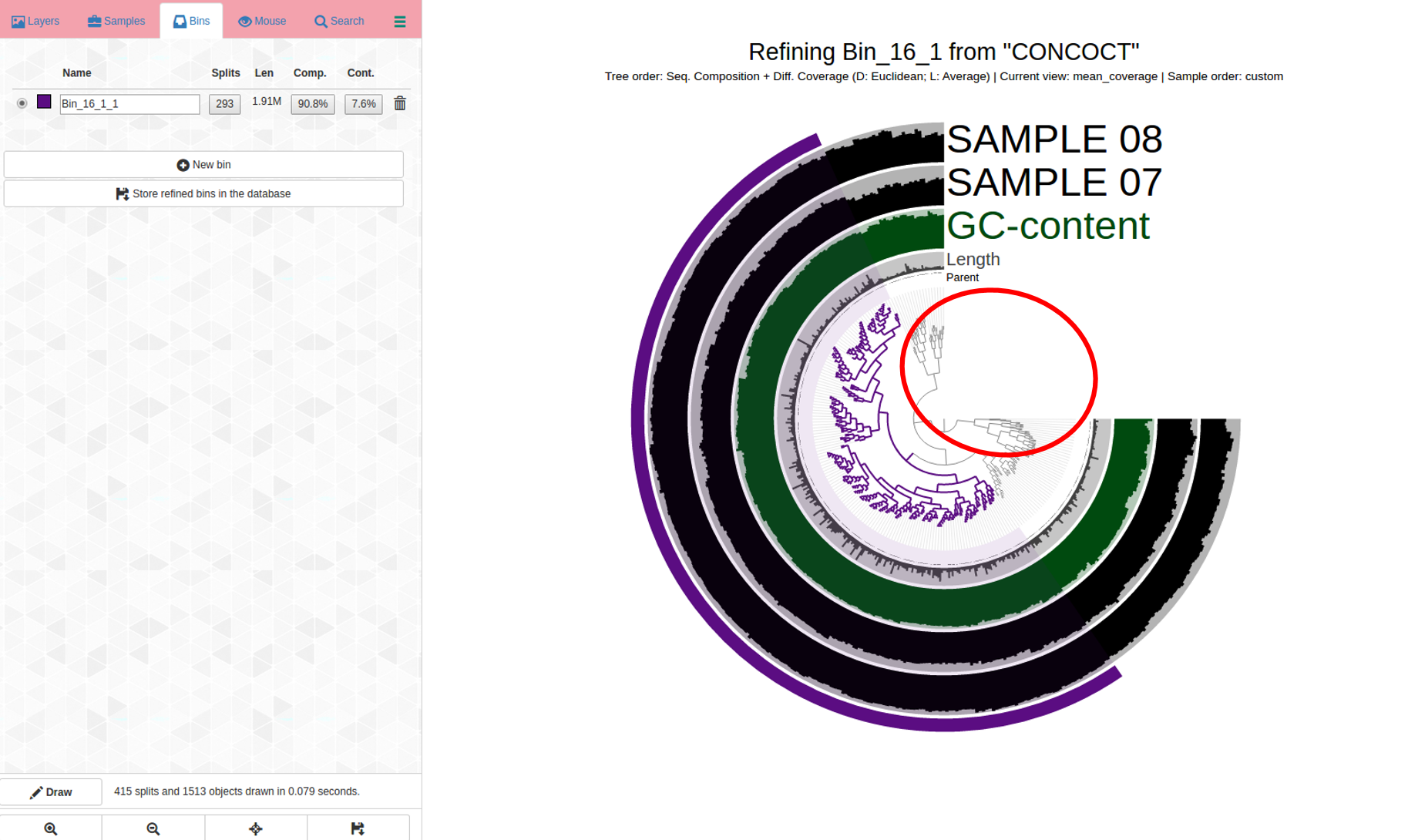
The bin is getting smaller (it is in purple now), it’s a work in progress. It is important to eliminate contigs that don’t belong; if completion percent goes down as well, that’s okay because it is obviously much more important to have a more “correct” bin than a mixed bin with high % completion.
Additionally, try to blast search several of the genes (right click on the interactive interface and the option comes up) for the outliers in the questionable part of the bin and the other parts and compare the taxonomy. For some samples, I can’t always do this (for example, marine sediment) because there might not be matches for any of it, so it depends on the system you are studying. Also, anvi’o has a feature that streamlines assigning taxonomy to contigs that you can include in your metagenomics workflow, which is described here
Circled in red: We can continue to refine this bin, eliminating outliers, which I use here to refer to contigs in the bin that are far- branching- the split in the dendrogram is very far so this is a sign those contigs might not belong; taken together with the coverage changing differently from one sample and the other, this is an indicator to consider removing the contigs; additionally, try re-drawing this with only sequence composition (drop-down option on interactive display). If some contigs are questionably assigned to a bin, I tend to err on the side of caution and remove them to get a more high-quality bin, even though it frequently means lowering the percent completion as well - that’s okay- quality over quantity.
A bin with very high redundancy: Bin_3 (C97/R86)
Lets look at a highly redundant bin, Bin_3 C97/R86:
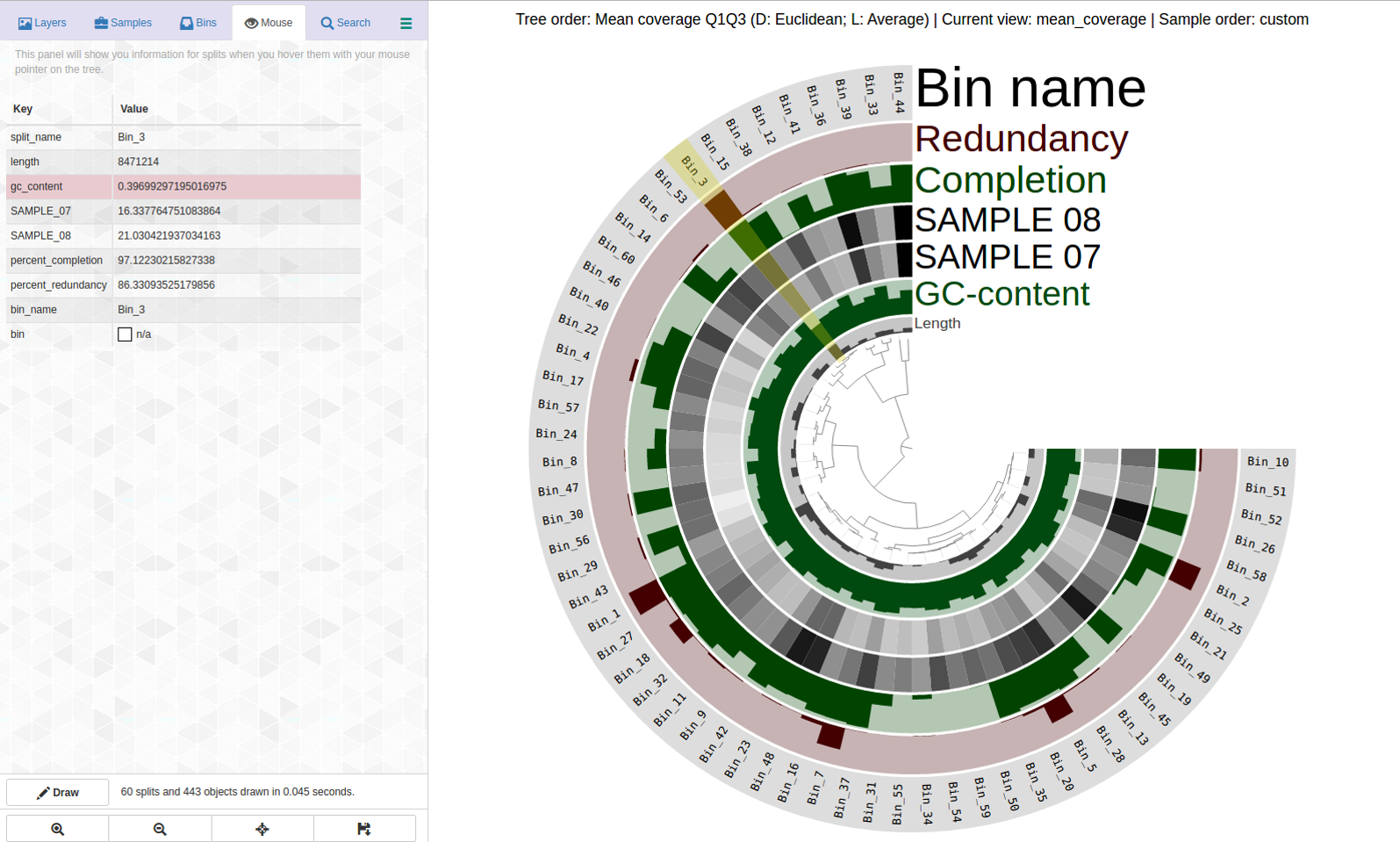
Can this be saved? This is what anvi-refine displays:
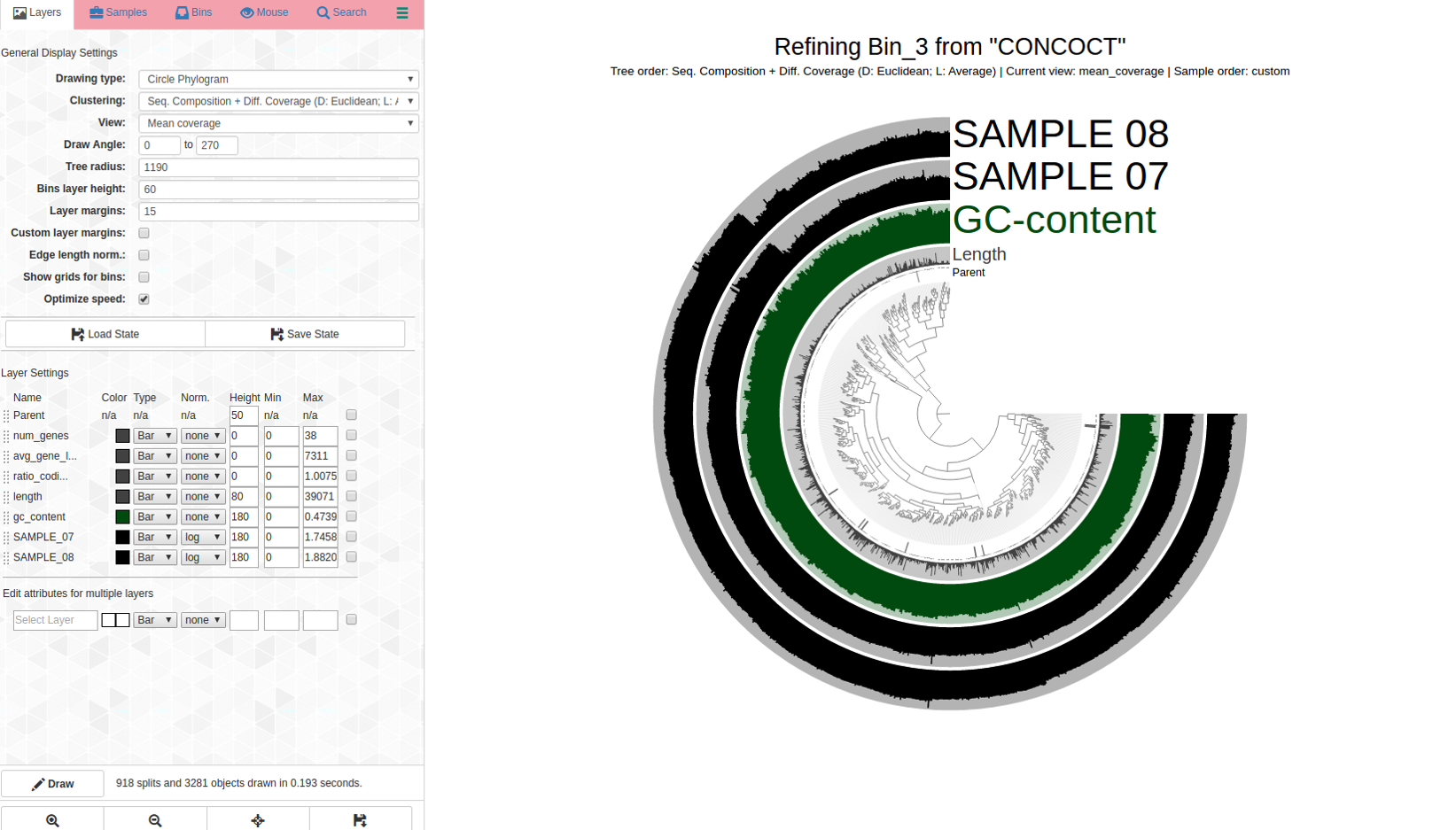
Ouch. There are dramatic differences in coverage and far branches, not surprising considering the high percent redundancy. Here is the first pass:
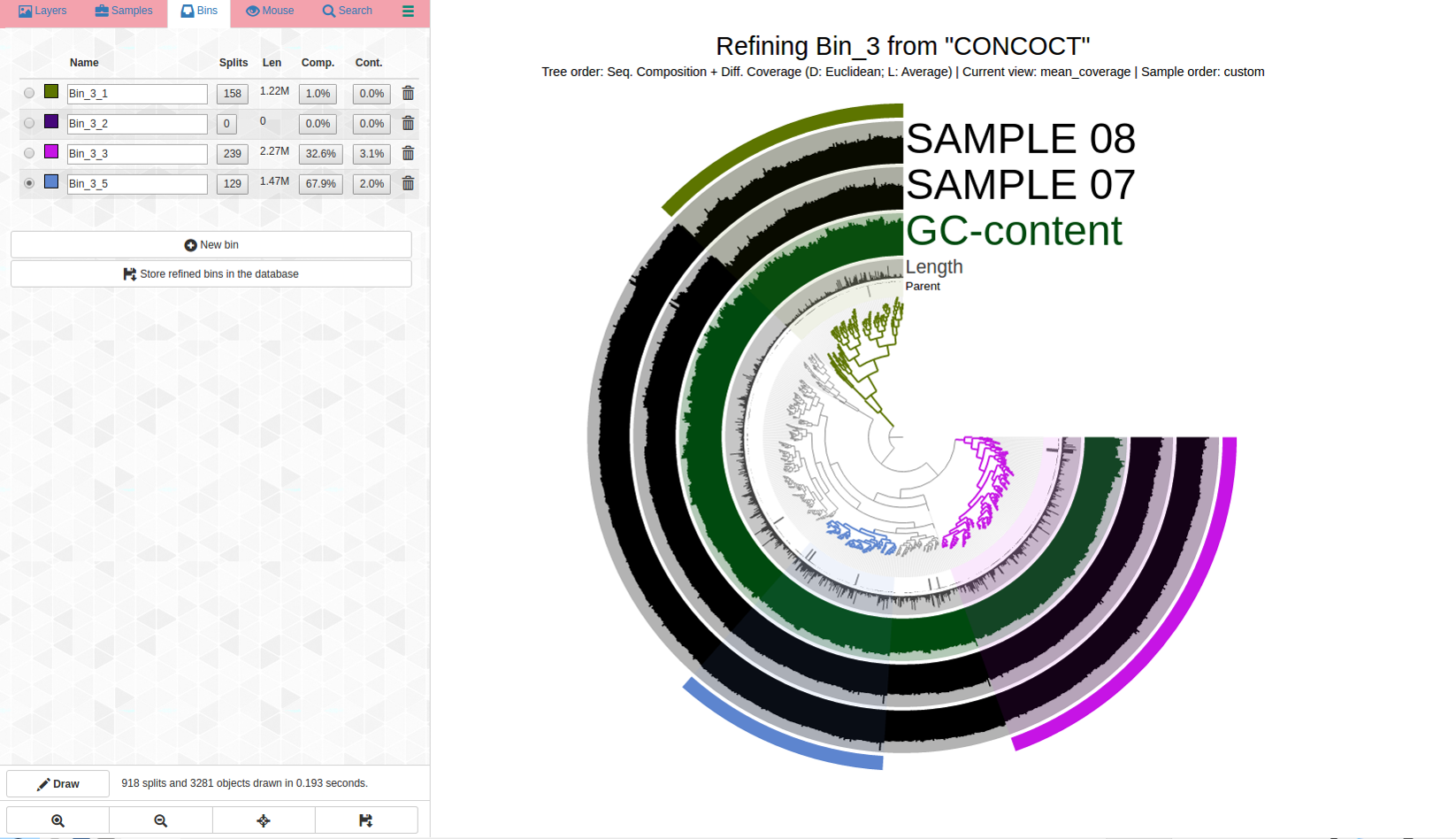
This is a very rough first pass at refining this very mixed bin, just to show what high-redundancy may look like. Bin 3-5, in blue, might be okay, although another look is needed. I included more contigs in Bin 3_5 (not shown), but the percent redundancy became much higher so that was worse, and that is why that bin is the size it is right now. So a lot of clicking is necessary to see what’s going on in the bin, and you have to explore what happens in several ways each time in order to inform your decision-making.
Save your selections with the “Store refined bins in the database” button in the bins panel, and open your new bins with anvi-refine again and take another look. By the way, the original bin is taken out of your database when you click “store refined bins” and is replaced with the new bin, so I saved an original version of my data before doing this (you can get a copy of your initial collection with the program anvi-export-collection so you can import it later).
Another bin with very good C/R still needs refinement
More indication that even for “good” bins, refinement may be necessary. Here is an example with Bin_35 with C97/R01 in my dataset:
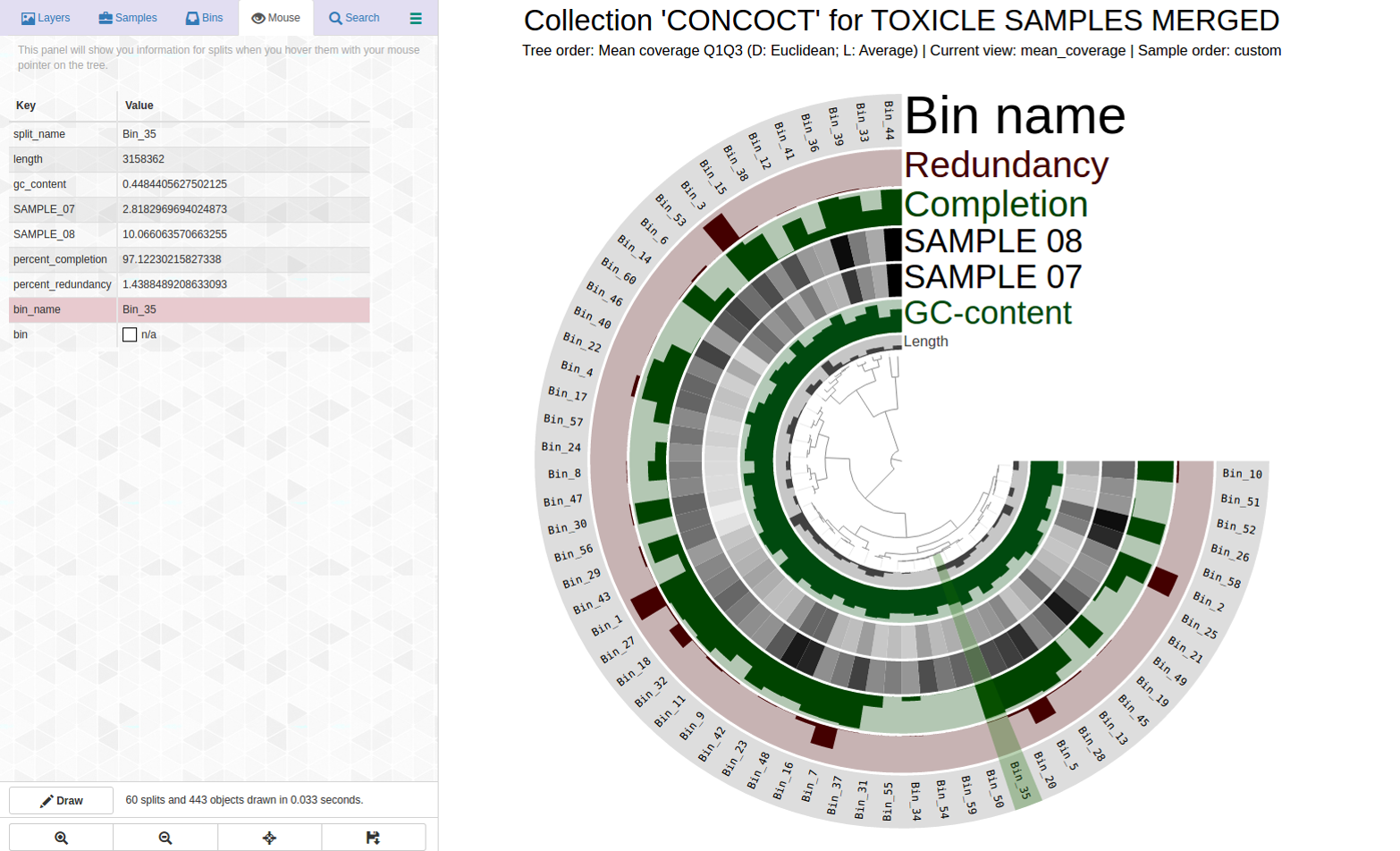
Here is the anvi-refine display for Bin_35:
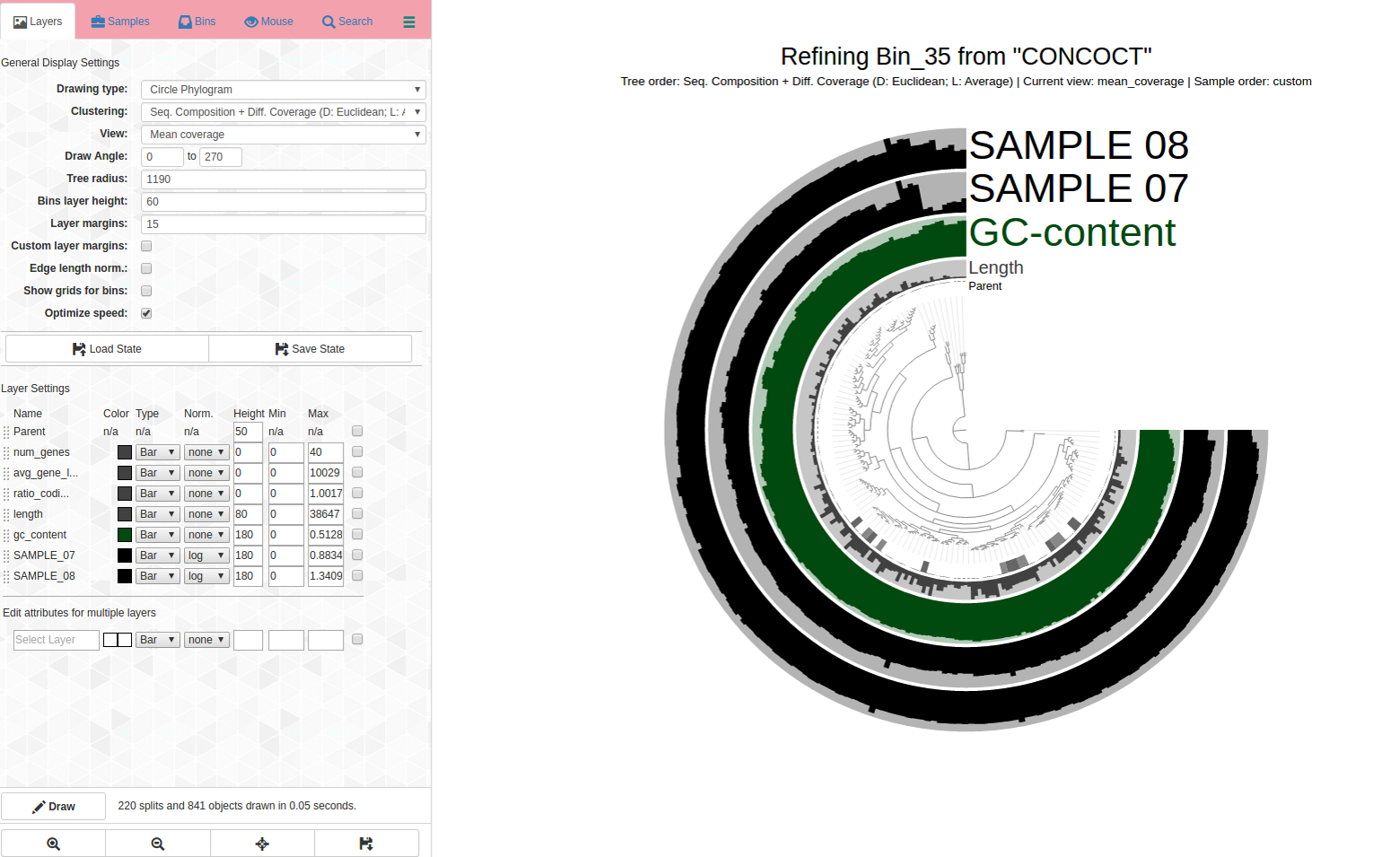
See the problems with how coverage is uneven and it is changing differently across samples, and how it branches? How would you refine this? See next image for my first pass refining:
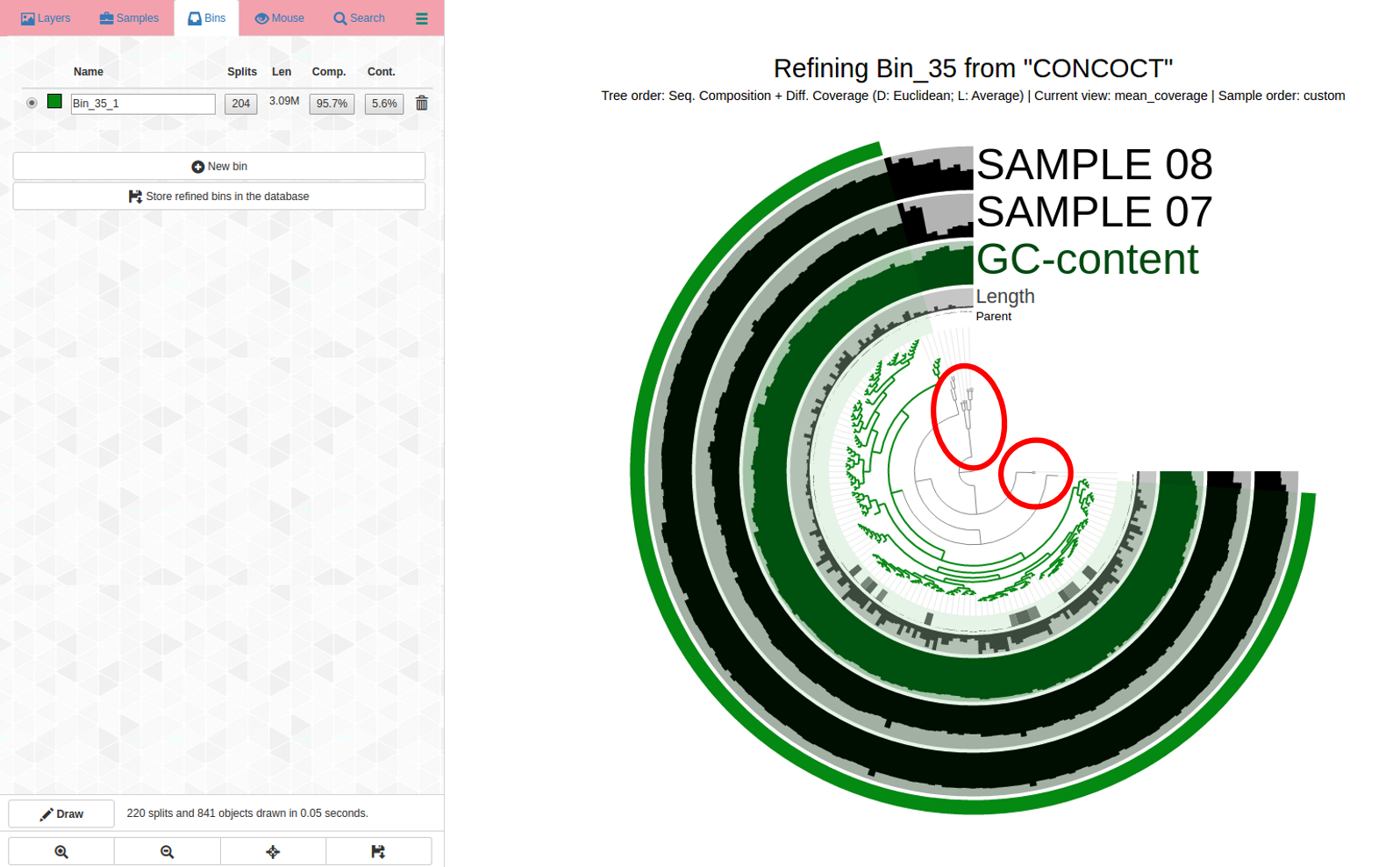
Circled in red: These are both outliers in terms of where they are in the dendrogram and the coverage changes are different for samples. It is a good idea to remove them from the bin to increase its purity. Yet another case refinement was important despite good C/R stats.
If you noticed that sometimes the C/R percents are slightly different in the interactive vs. the refine screen- this is because it depends on whether you are looking at the average of the single copy gene collections or only one of them.
Yet another highly redundant bin
Let’s take a look at the highly redundant Bin_1 (C100/R81):
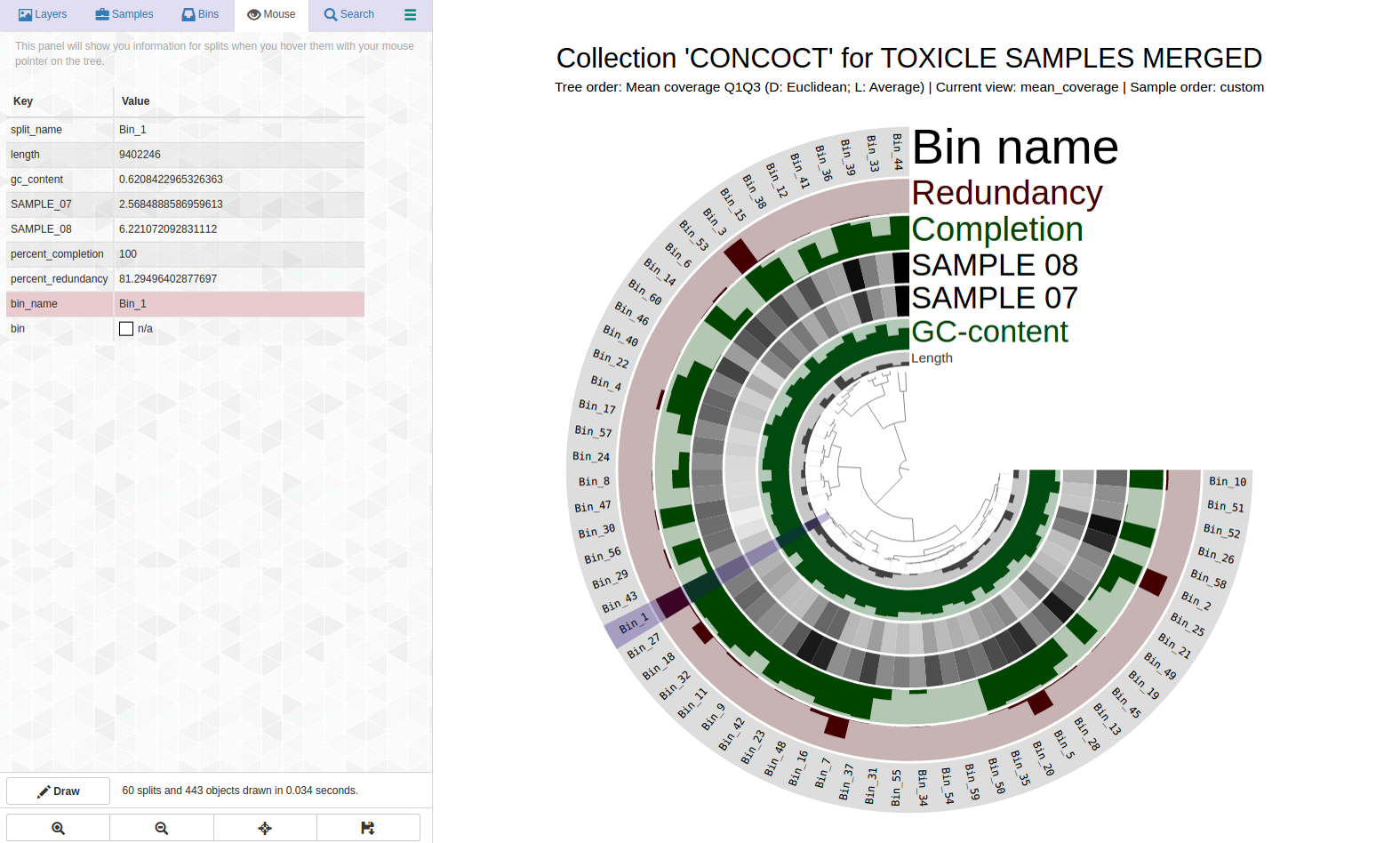
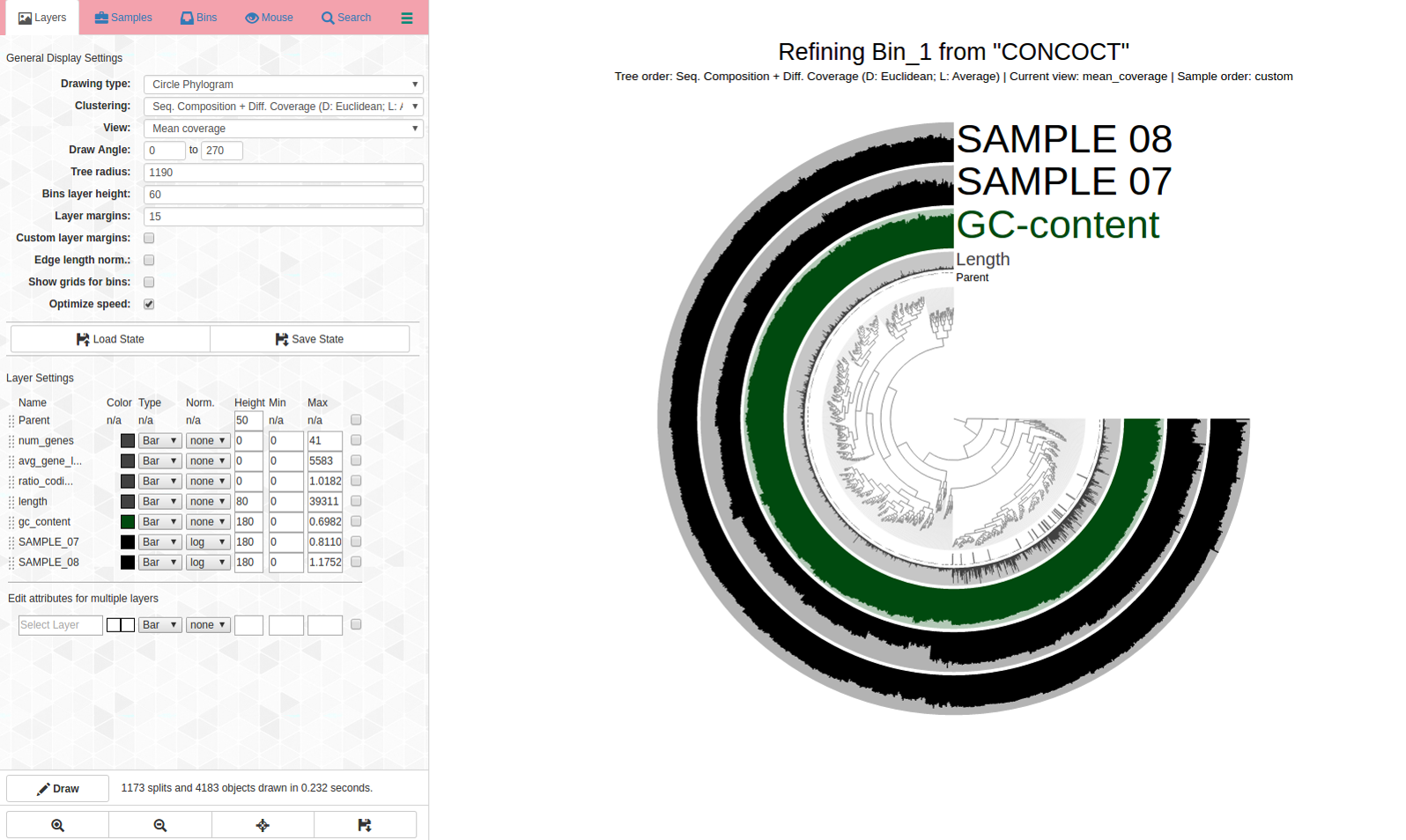
Can we pull out a bin or two from this? Here is the anvi-refine display:
And here are my selections, Bin_1_1 (C75/R6.4), and Bin_1_2 (C98/R6.0):
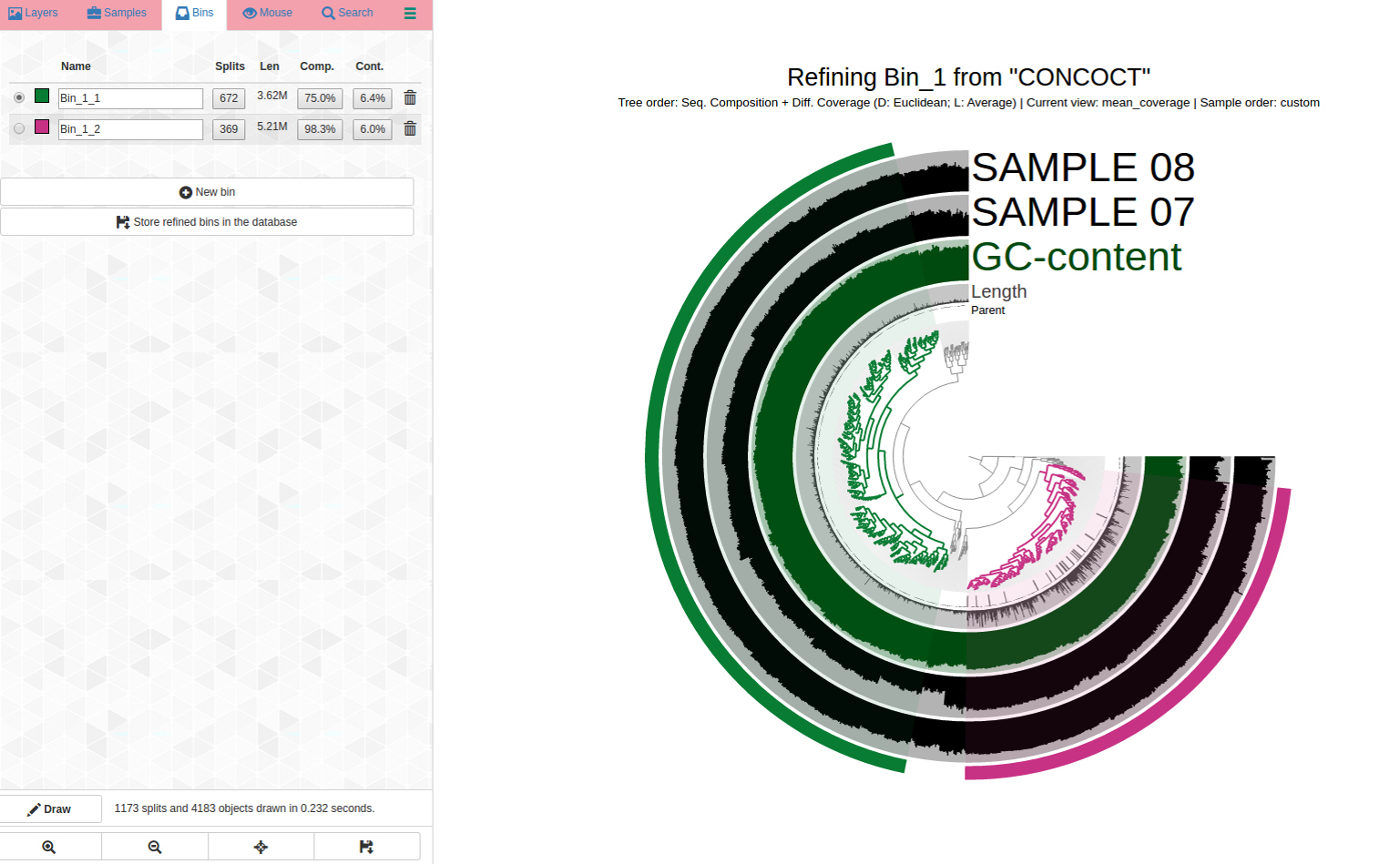
This is how manual refinement can untangle initially mixed bins.
Conclusion
Anvi’o is incredibly useful for genome refinement, which is crucial for working with MAGs.