
Table of Contents
If you are using the phylogenomics workflow with anvi’o version v5.1 with internal genomes, you must read this issue report as this will have a significant impact your findings.
The latest version of anvi’o is v7.1. See the release notes.
This tutorial is tailored for anvi’o v4 or later. You can learn which version you have on your computer by typing anvi-profile --version in your terminal.
The goal of this tutorial is to walk you through some of the anvi’o capabilities that can help you study your data with phylogenomics. With the current anvi’o phylogenomics workflow,
-
You can run phylogenomic analyses on a bunch of FASTA files, on your metagenome-assembled genomes stored in an anvi’o collection, or on a combination of the two by combining your MAGs with genomes from other resources.
-
You can use any set of genes to create concatenated proteins. While you can use genes in one of the default HMM profiles anvi’o uses for bacterial and archaeal single-copy core genes (such as a set of commonly used ribosomal proteins), you can also go rogue and use a set of genes in your custom HMM profile.
-
You can combine pangenomics and phylogenomics to investigate relationships between very closely related genomes using gene clusters beyond universal single-copy core genes.
-
You can interactively visualize your phylogenomic trees using the anvi’o interactive interface (by optionally extending them with additional data), or share them with your colleagues through anvi’server.
Please consider reading the entire document even if you are planning to use only anvi’o / phylogenomics only for your pangenomes, or only for your MAGs. There are notes that apply to both, yet mentioned only once throughout the document to minimize redundancy.
Preface
What is phylogenomics?
Well, maybe to some people it is a guy on Twitter (cough who happened to write this seminal paper and coined the term cough), while maybe to some others it is a way to make all those already-unreliable trees even worse by putting the concept of phylogenetics on steroids. But I personally quite like the Wikipedia definition on it:
Phylogenomics is the intersection of the fields of evolution and genomics. The term has been used in multiple ways to refer to analysis that involves genome data and evolutionary reconstructions. It is a group of techniques within the larger fields of phylogenetics and genomics. Phylogenomics draws information by comparing entire genomes, or at least large portions of genomes. Phylogenetics compares and analyzes the sequences of single genes, or a small number of genes, as well as many other types of data.
Phylogenomics is not only useful to make sense of the diversity of life (as Hug et al. beautifully did it here), but it is also quite relevant to much more practical needs of the age of high-throughput recovery of single-cell, isolate, and metagenome-assembled genomes.
The anvi’o solution explained in this tutorial to address this theoretical and practical need is neither the first nor the only convenient solution. For instance, you could use PhyloSift to infer phylogenomic relationships between multiple genomes, which is a developed by Darling and his colleagues. Aaron Darling is cool like this, and we have been using PhyloSift in our lab happily. So all good. While PhyloSift is very convenient, and will be enough for the vast majority of scientists as it automatically identifies a set of marker genes in genomes of interest, anvi’o gives you full control over the phylogenomic workflow by letting you pick the genes for your analysis, offering more room for activities (which may quickly turn into a ‘losing our mental health rapidly’ experience depending on how you do the science thing, and how deep you are willing to dive into your data).
Example data pack
If you want to follow this tutorial with the data I used here, you can type the following commands to download the data pack and set the stage:
# download the data pack
wget https://ndownloader.figshare.com/files/28715136 -O AnvioPhylogenomicsTutorialDataPack.tar.gz
# unpack it
tar -zxvf AnvioPhylogenomicsTutorialDataPack.tar.gz
# go into the directory
cd AnvioPhylogenomicsTutorialDataPack
# take a peak
$ ls AnvioPhylogenomicsTutorialDataPack
closely-related distantly-related
Now we can start.
Working with metagenome-assembled genomes stored in anvi’o collections
Indeed the phylogenomic workflow natively integrated with everything else in anvi’o, and you can use it to work with your bins in collections stored in anvi’o profiles. But to keep this tutorial as abstract as possible, I will outsource examples using anvi’o profiles to the anvi’o tutorial on the Infant Gut Dataset, in which the use of phylogenomics is demonstrated in detail right at this section. Please don’t hesitate to ask for help if you are here and need more information. We will happily expand this section if necessary.
Working with FASTA files
This part of the tutorial is taking place in the data pack directory distantly-related
Let’s assume you have a number of FASTA files –like the ones I randomly chose from multiple taxa:
$ ls
Bacteroides_fragilis_2334.fa Bacteroides_fragilis_2347.fa
Escherichia_coli_6920.fa Prevotella_dentalis_19591.fa
Prevotella_intermedia_19600.fa Salmonella_enterica_22047.fa
Bacteroides_fragilis_2346.fa Escherichia_albertii_6917.fa
Escherichia_coli_9038.fa Prevotella_denticola_19594.fa
Salmonella_enterica_21806.fa Salmonella_enterica_22289.fa
The first thing we need to do is to generate an anvi’o contigs database for each one of them:
for i in `ls *fa | awk 'BEGIN{FS=".fa"}{print $1}'`
do
anvi-gen-contigs-database -f $i.fa -o $i.db -T 4
anvi-run-hmms -c $i.db
done
At the end of this, for each FASTA file I should have a file with the same name that ends with ‘.db’.
The next step is to define all the contigs databases of interest, and give them a name to introduce them to anvi’o. Let’s call this file ‘external-genomes.txt’:
| name | contigs_db_path |
|---|---|
| Bacteroides_fragilis_2334 | Bacteroides_fragilis_2334.db |
| Bacteroides_fragilis_2346 | Bacteroides_fragilis_2346.db |
| Bacteroides_fragilis_2347 | Bacteroides_fragilis_2347.db |
| Escherichia_albertii_6917 | Escherichia_albertii_6917.db |
| Escherichia_coli_6920 | Escherichia_coli_6920.db |
| Escherichia_coli_9038 | Escherichia_coli_9038.db |
| Prevotella_dentalis_19591 | Prevotella_dentalis_19591.db |
| Prevotella_denticola_19594 | Prevotella_denticola_19594.db |
| Prevotella_intermedia_19600 | Prevotella_intermedia_19600.db |
| Salmonella_enterica_21806 | Salmonella_enterica_21806.db |
| Salmonella_enterica_22047 | Salmonella_enterica_22047.db |
| Salmonella_enterica_22289 | Salmonella_enterica_22289.db |
You can download this file into your work directory:
wget https://goo.gl/XuezQF -O external-genomes.txt
We will use the program anvi-get-sequences-for-hmm-hits to get sequences out of this collection of contigs databases.
anvi-get-sequences-for-hmm-hits is quite a powerful program that lets you do lots of things with a single anvi’o contigs database, or a collection of external genomes (which is what we are doing here), or a collection in a profile database. Please consider exploring it yourself since this tutorial will not cover all aspects of it.
Fine. We will concatenate genes, but which genes are we going to concatenate?
First, we need to identify an HMM profile to use, and then select some gene names from this profile to play with. Anvi’o has multiple HMM profiles for single-copy core genes by default, and you can always use the program anvi-db-info to learn which HMM sources you have in a given contigs database. Here is an example:
anvi-db-info Salmonella_enterica_21170.db
(...)
AVAILABLE HMM SOURCES
===============================================
* 'Archaea_76' (76 models with 37 hits)
* 'Bacteria_71' (71 models with 70 hits)
* 'Protista_83' (83 models with 5 hits)
* 'Ribosomal_RNA_12S' (1 model with 0 hits)
* 'Ribosomal_RNA_16S' (3 models with 7 hits)
* 'Ribosomal_RNA_18S' (1 model with 0 hits)
* 'Ribosomal_RNA_23S' (2 models with 7 hits)
* 'Ribosomal_RNA_28S' (1 model with 0 hits)
* 'Ribosomal_RNA_5S' (5 models with 0 hits)
For the remainder of this tutorial we will use the collection called Bacteria_71, which is the default bacterial single-copy core gene collection anvi’o (which also includes ribosomal proteins that are good for archaea as well). However, you can use others, including your own custom HMM profiles to focus on a set of single-copy genes across your genomes of interest (for instance, if you are comparing multiple eukaryotic genomes, neither of these collections may be very useful to you).
OK, so we will use Bacteria_71 as a source of single-copy core genes, but which genes exactly should we pick from it? Let’s take a look and see which genes are described in this collection:
$ anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt \
--hmm-source Bacteria_71 \
--list-available-gene-names
Internal genomes .............................: 0 have been initialized.
External genomes .............................: 12 found.
GENES IN HMM SOURCES COMMON TO ALL 12 GENOMES
===============================================
* Bacteria_71 [type: singlecopy]: ADK, AICARFT_IMPCHas, ATP-synt, ATP-synt_A,
Adenylsucc_synt, Chorismate_synt, EF_TS, Exonuc_VII_L, GrpE, Ham1p_like, IPPT,
OSCP, PGK, Pept_tRNA_hydro, RBFA, RNA_pol_L, RNA_pol_Rpb6, RRF, RecO_C,
Ribonuclease_P, Ribosom_S12_S23, Ribosomal_L1, Ribosomal_L13, Ribosomal_L14,
Ribosomal_L16, Ribosomal_L17, Ribosomal_L18p, Ribosomal_L19, Ribosomal_L2,
Ribosomal_L20, Ribosomal_L21p, Ribosomal_L22, Ribosomal_L23, Ribosomal_L27,
Ribosomal_L27A, Ribosomal_L28, Ribosomal_L29, Ribosomal_L3, Ribosomal_L32p,
Ribosomal_L35p, Ribosomal_L4, Ribosomal_L5, Ribosomal_L6, Ribosomal_L9_C,
Ribosomal_S10, Ribosomal_S11, Ribosomal_S13, Ribosomal_S15, Ribosomal_S16,
Ribosomal_S17, Ribosomal_S19, Ribosomal_S2, Ribosomal_S20p, Ribosomal_S3_C,
Ribosomal_S6, Ribosomal_S7, Ribosomal_S8, Ribosomal_S9, RsfS, RuvX, SecE, SecG, SecY, SmpB, TsaE, UPF0054, YajC, eIF-1a, ribosomal_L24, tRNA-synt_1d,
tRNA_m1G_MT
You can select any combination of these genes to use for your phylogenomic analysis, including all of them –if you do not declare a --gene-names parameter, the program would use all. But considering the fact that ribosomal proteins are often used for phylogenomic analyses, let’s say we decided to use the following ribosomal proteins: Ribosomal_L1, Ribosomal_L2, Ribosomal_L3, Ribosomal_L4, Ribosomal_L5, and Ribosomal_L6.
The following command will give you the concatenated amino acid sequences for these genes:
anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt \
-o concatenated-proteins.fa \
--hmm-source Bacteria_71 \
--gene-names Ribosomal_L1,Ribosomal_L2,Ribosomal_L3,Ribosomal_L4,Ribosomal_L5,Ribosomal_L6 \
--return-best-hit \
--get-aa-sequences \
--concatenate
Please examine the resulting FASTA file concatenated-proteins.fa to better understand what just happened.
Special Thanks
Until very recently, the program anvi-get-sequences-for-hmm-hits did not have a --concatenate flag. It could report sequences for HMM hits, but it would not generate concatenated gene alignments for users to perform phylogenomic analyses.
We finally implemented this, because Luke McKay asked for it in an e-mail he sent to the anvi’o discussion group.
There is only one thing more valuable than getting pushed by the community, which in return shapes our research back in our lab, and that is chocolate. Luke had visited our lab recently, and had sent back some Moose Drool and chocolate from Montana:

Of course there was no way we were not going to implement the --concatenate flag ;) Thank you, Luke!
Now we have the FASTA file, we can use the program anvi-gen-phylogenomic-tree,
anvi-gen-phylogenomic-tree -f concatenated-proteins.fa \
-o phylogenomic-tree.txt
to get a newick-formatted phylogeny artifact for our genomes!
Important note: FastTree is currently the only option implemented in anvi-gen-phylogenomic-tree. Its purpose is to make sure you have a quick and easy way to perform your phylogenomic analyses, but you have the freedom (or responsibility, depending on how you see things) to try other means to get your trees. Some of the options we are familiar with include MrBayes, MEGA, and PHYLIP, among many others. We typically use XX, and you can go through this workflow to get yourself familiarized with how we use it. Luckily, almost all of the programs you will find to generate trees are going to be happy to work with the concatenated amino acid sequences you will get from anvi’o. Phylogenetics has tremendous challenges, investigated by hundreds of brilliant scientists over decades. And here we are, reducing years of research into a single command line. We are doing this, because at some point we have to stop and be practical, but serious questions and good data always deserve more. You can get away by using one algorithm to generate your tree, but you should consider exploring other options. If you happen to know a good written resource, or if you are a good resource, please write to us, and help us think about what would be the best strategy to provide useful insights to researchers who may be interested in learning more about the details of phylogenetics and/or phylogenomics.
Now you have a newick tree that shows you how your genomes relate to each other, and you can use the anvi’o interactive interface to immediately visualize this new tree:
In manual mode, anvi’o will automatically generate an empty profile database upon its first initialization. For instance, even though you do not have the profile database phylogenomic-profile.db in your work directory if you are following the tutorial, when you run the command below, it will be automatically generated.
anvi-interactive -p phylogenomic-profile.db \
-t phylogenomic-tree.txt \
--title "Phylogenomics Tutorial Example #1" \
--manual
and play with the settings a bit (here is an interactive version):
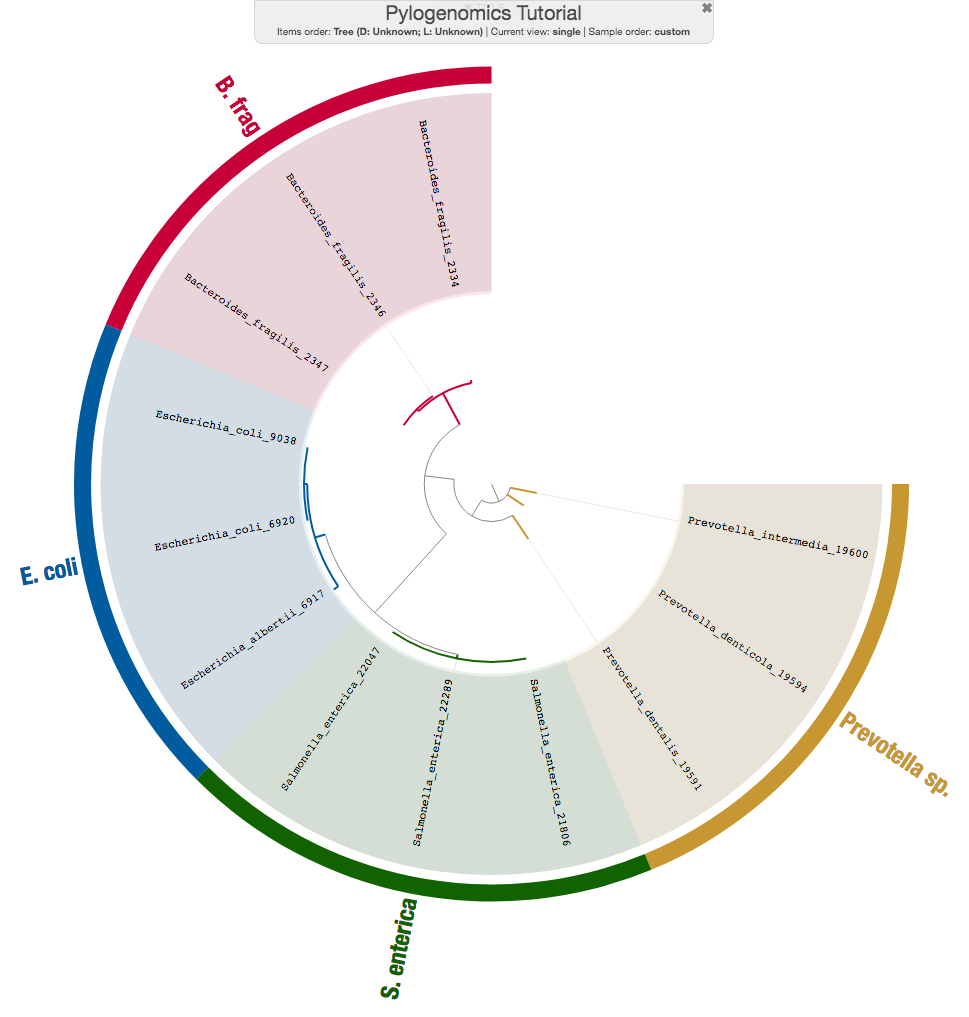
You can learn much more about the --manual mode of the anvi-interactive here: A tutorial on the anvi’o interactive interface.
The trees you will get from FastTree will not be rooted (including the tree you see in the figure above), and you should consider rooting them. FigTree is a great software for this and other tree operations. After rooting your tree, you can come back to anvi’o with the new newick file, or go somewhere else (in which case we you are not allowed to use anvi’o ever again, of course, and thankyouverymuch (Anvi’o developers hope that you read the last sentence in Frank Underwood’s voice)).
It is also possible to extend the display with additional information. For instance, we did this for the phylogenomics of some of the MAGs we recovered from TARA Oceans. Let’s first assume these additional data for the 12 genomes we have:
| genome_id | genome | phylum | genus |
|---|---|---|---|
| Bacteroides_fragilis_2334 | B. fragilis 03 | Bacteroidetes | Bacteroides |
| Bacteroides_fragilis_2346 | B. fragilis 02 | Bacteroidetes | Bacteroides |
| Bacteroides_fragilis_2347 | B. fragilis 01 | Bacteroidetes | Bacteroides |
| Escherichia_albertii_6917 | E. albertii | Proteobacteria | Escherichia |
| Escherichia_coli_6920 | E. coli 02 | Proteobacteria | Escherichia |
| Escherichia_coli_9038 | E. coli 01 | Proteobacteria | Escherichia |
| Prevotella_dentalis_19591 | P. dentalis | Bacteroidetes | Prevotella |
| Prevotella_denticola_19594 | P. denticola | Bacteroidetes | Prevotella |
| Prevotella_intermedia_19600 | P. intermedia | Bacteroidetes | Prevotella |
| Salmonella_enterica_21806 | S. enterica 01 | Proteobacteria | Salmonella |
| Salmonella_enterica_22047 | S. enterica 02 | Proteobacteria | Salmonella |
| Salmonella_enterica_22289 | S. enterica 03 | Proteobacteria | Salmonella |
You can download this file into your work directory:
wget https://goo.gl/UZDbC8 -O view.txt
Now you can run the interactive interface again to see the additional layers:
anvi-interactive -p phylogenomic-profile.db \
-d view.txt \
-t phylogenomic-tree.txt \
--title "Phylogenomics Tutorial Example #2" \
--manual
This is my (interactive) version after a bit of tinkering with colors:
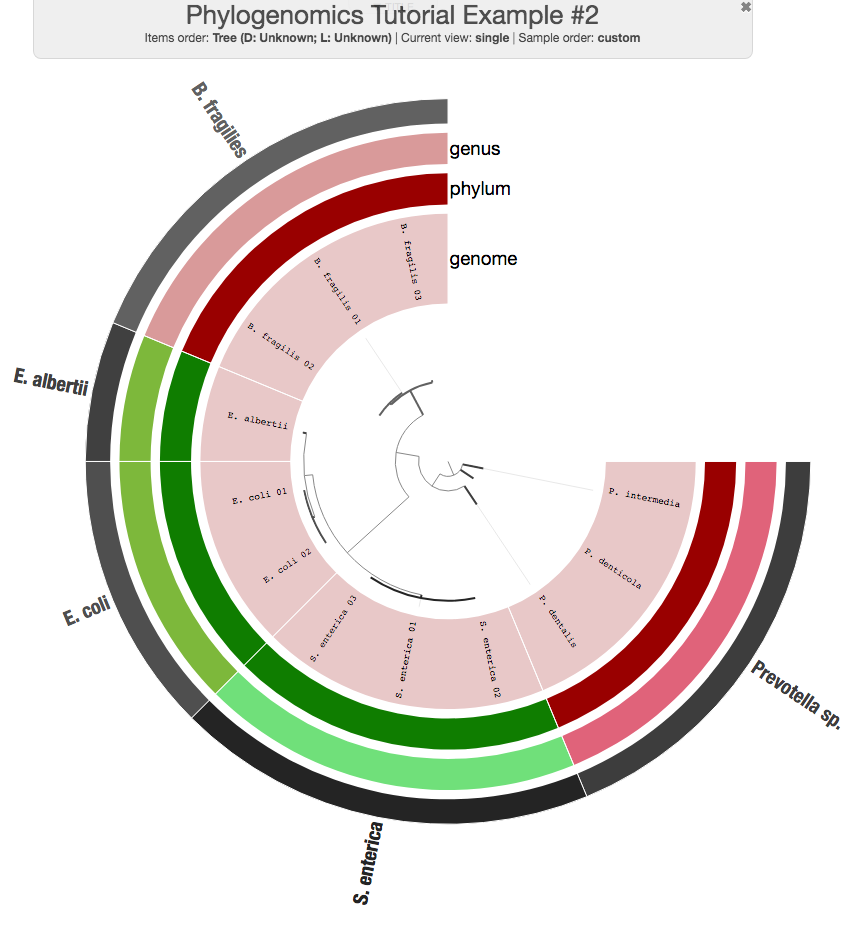
anvi-get-sequences-for-hmm-hits will also accept the gene names of interest in a file if you provide a file path to the parameter --gene-names.
For instance, instead of the 6 ribosomal proteins we used so far in this tutorial, one could have used all the ribosomal proteins described in Bacteria_71, which would have been very unpleasant to try to fit into a single command line as comma-separated names.
To demonstrate that, I put all those 39 ribosomal proteins into a file:
$ cat gene-names.txt
Ribosomal_L1
Ribosomal_L2
Ribosomal_L13
Ribosomal_L14
Ribosomal_L16
Ribosomal_L17
Ribosomal_L18p
Ribosomal_L19
Ribosomal_L20
Ribosomal_L21p
Ribosomal_L22
Ribosomal_L23
ribosomal_L24
Ribosomal_L27
Ribosomal_L27A
Ribosomal_L28
Ribosomal_L29
Ribosomal_L3
Ribosomal_L32p
Ribosomal_L35p
Ribosomal_L4
Ribosomal_L5
Ribosomal_L6
Ribosomal_L9_C
Ribosomal_S10
Ribosomal_S11
Ribosomal_S13
Ribosomal_S15
Ribosomal_S16
Ribosomal_S17
Ribosomal_S19
Ribosomal_S2
Ribosomal_S3_C
Ribosomal_S6
Ribosomal_S7
Ribosomal_S8
Ribosomal_S9
Ribosomal_S20p
Ribosom_S12_S23
And re-run the anvi’o analysis with this file:
# get the gene sequences using the file
anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt \
-o concatenated-ribosomal-proteins.fa \
--hmm-source Bacteria_71 \
--gene-names gene-names.txt \
--return-best-hit \
--get-aa-sequences \
--concatenate
# compute the phylogenomic tree
anvi-gen-phylogenomic-tree -f concatenated-ribosomal-proteins.fa \
-o phylogenomic-ribosomal-tree.txt
So now we have a new tree.
While we are at it, Tom also run PhyloSift on these genomes just to make sure the results from our workflow does make sense:
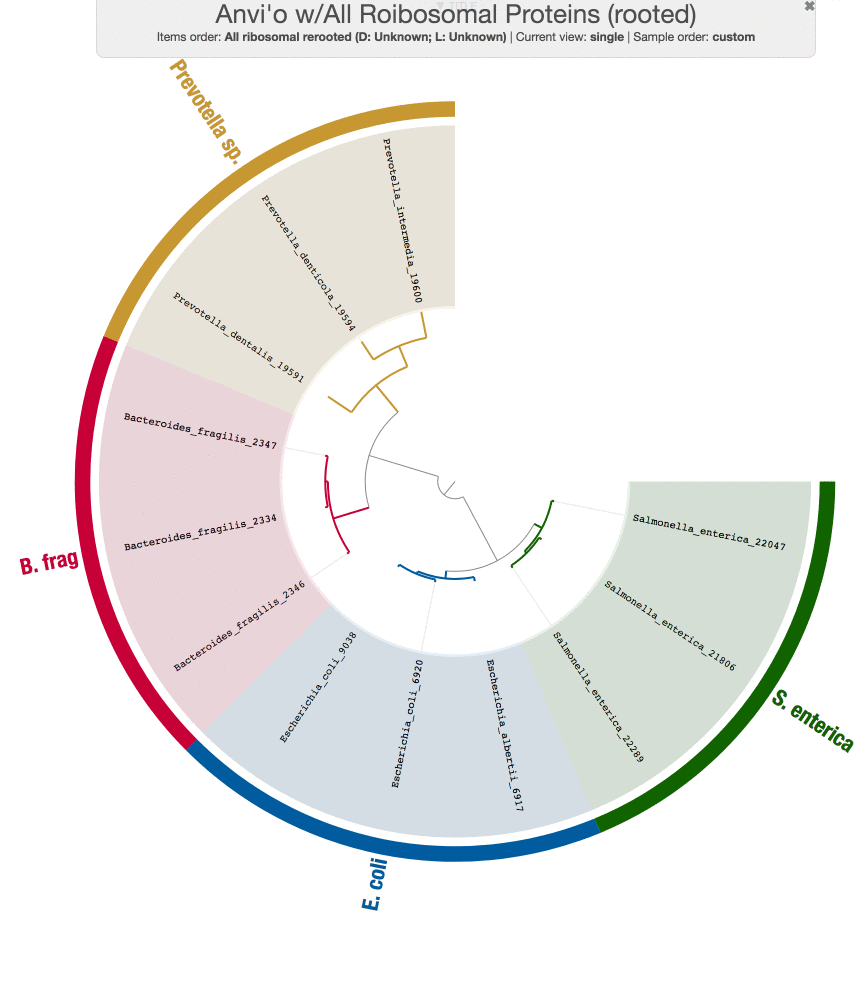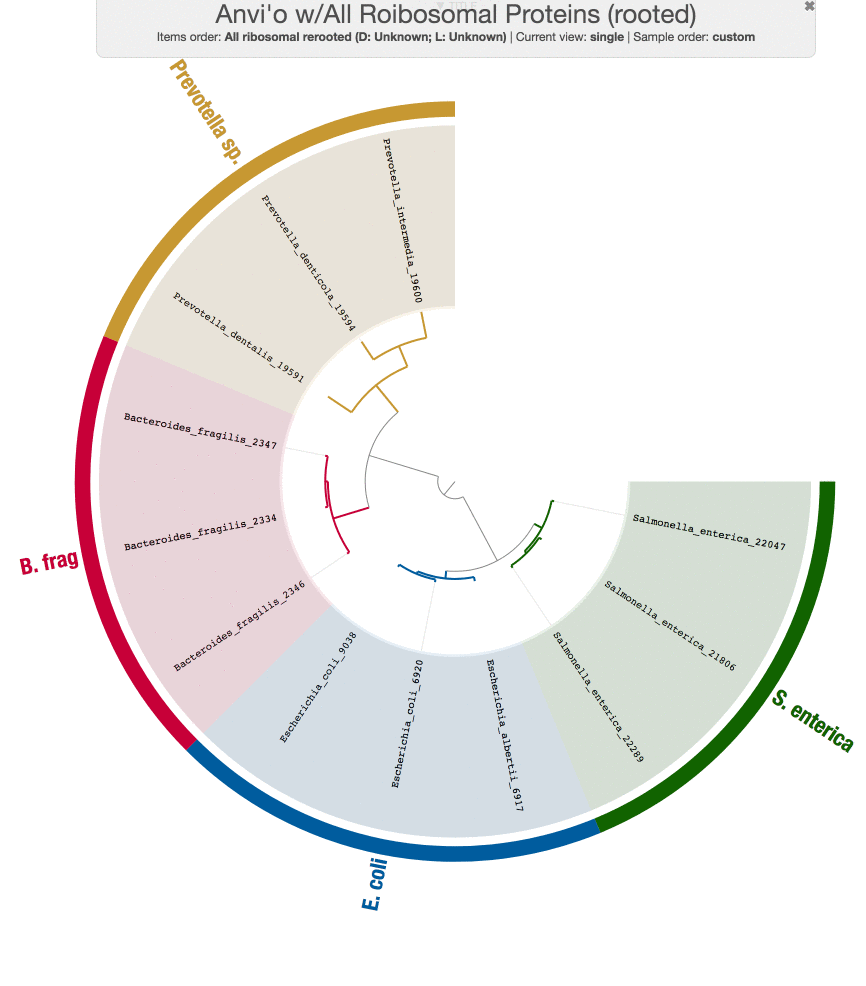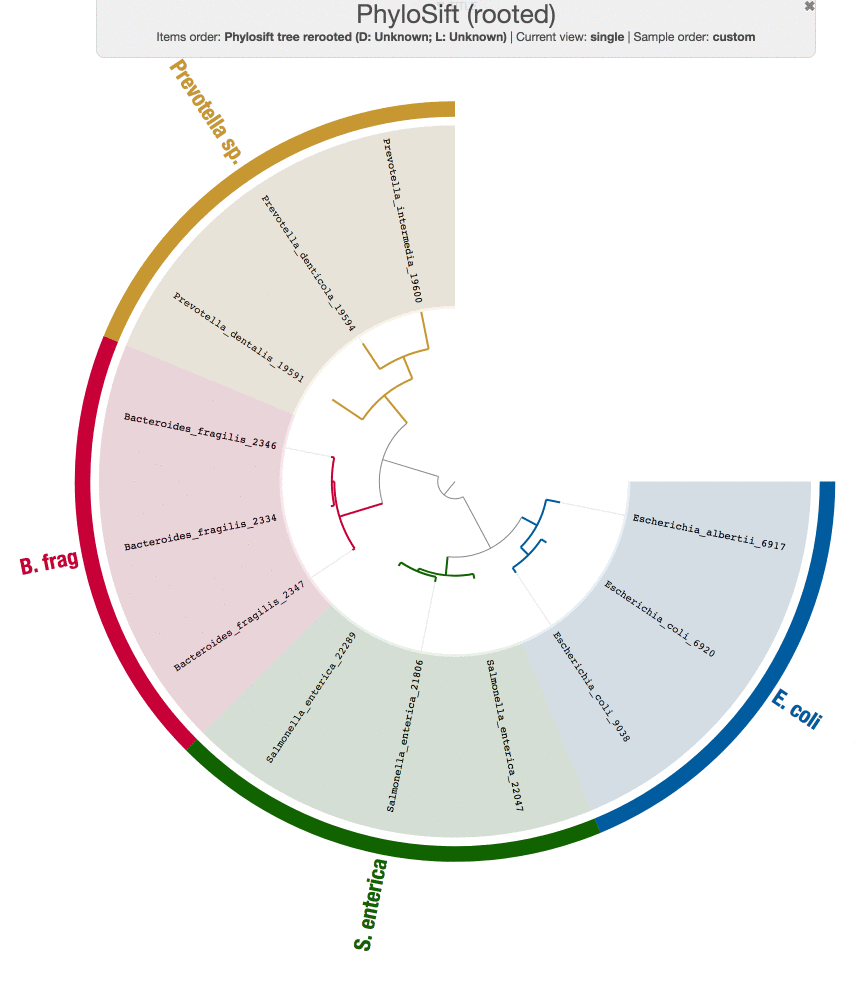
Both tree structures are very comparable, and small differences are likely coming from the fact that we didn’t use the same set of genes PhyloSift uses.
Working with gene clusters in anvi’o pangenomes
This part of the tutorial is taking place in the data pack directory closely-related
Let’s assume we have the following genomes:
$ ls
Salmonella_enterica_21170.fa Salmonella_enterica_22024.fa
Salmonella_enterica_21496.fa Salmonella_enterica_22047.fa
Salmonella_enterica_21642.fa Salmonella_enterica_22258.fa
Salmonella_enterica_21805.fa Salmonella_enterica_22289.fa
Salmonella_enterica_21806.fa Salmonella_enterica_24368.fa
Salmonella_enterica_21834.fa Salmonella_enterica_24443.fa
As it is discussed in the previous section, to do anything with these genomes, we will need them in the anvi’o contigs database format, so let’s start with that:
for i in `ls *fa | awk 'BEGIN{FS=".fa"}{print $1}'`
do
anvi-gen-contigs-database -f $i.fa -o $i.db
anvi-run-hmms -c $i.db
done
At the end of this the directory should be populated with files that ends with ‘.db’, but we are not done: we also need a TAB-delimited ‘external genomes’ file to describe these genomes and associate them with a name. Here is mine:
| name | contig_db_path |
|---|---|
| Salmonella_enterica_21170.db | Salmonella_enterica_21170.db |
| Salmonella_enterica_21496.db | Salmonella_enterica_21496.db |
| Salmonella_enterica_21642.db | Salmonella_enterica_21642.db |
| Salmonella_enterica_21805.db | Salmonella_enterica_21805.db |
| Salmonella_enterica_21806.db | Salmonella_enterica_21806.db |
| Salmonella_enterica_21834.db | Salmonella_enterica_21834.db |
| Salmonella_enterica_22024.db | Salmonella_enterica_22024.db |
| Salmonella_enterica_22047.db | Salmonella_enterica_22047.db |
| Salmonella_enterica_22258.db | Salmonella_enterica_22258.db |
| Salmonella_enterica_22289.db | Salmonella_enterica_22289.db |
| Salmonella_enterica_24368.db | Salmonella_enterica_24368.db |
| Salmonella_enterica_24443.db | Salmonella_enterica_24443.db |
Which you can download into your work directory this way:
wget https://goo.gl/DTM9sz -O external-genomes.txt
Before we start, there is a question you may be asking yourself: why don’t we analyze these genomes the way it is explained in the previous section?
Why gene clusters?
What makes single-copy core genes powerful for some applications of phylogenomics, makes them weak for other applications. We use single-copy core genes because we find them in most of the bacterial and archaeal genomes. However, comparing very similar genomes may require the inclusion of a larger number of genes that occur in all genomes of interest to increase the signal of divergence.
For instance, let’s quickly run the first method:
# get the gene sequences
anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt \
-o concatenated-proteins.fa \
--hmm-source Bacteria_71 \
--gene-names Ribosomal_L1,Ribosomal_L2,Ribosomal_L3,Ribosomal_L4,Ribosomal_L5,Ribosomal_L6 \
--return-best-hit \
--get-aa-sequences \
--concatenate
# compute the phylogenomic tree
anvi-gen-phylogenomic-tree -f concatenated-proteins.fa \
-o phylogenomic-tree.txt
# run the interactive interface
anvi-interactive -p phylogenomic-profile.db \
-t phylogenomic-tree.txt \
--title "Phylogenomics Tutorial Example #3" \
--manual
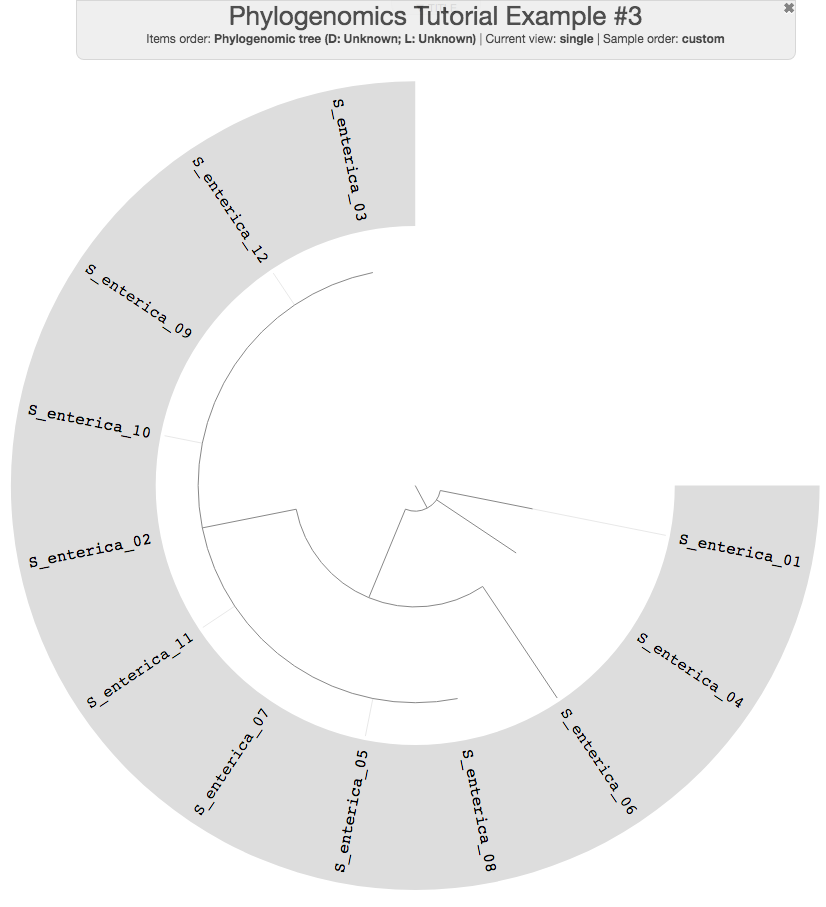
As you can see, 9 of the genomes have a flat line in the phylogenomic tree, which indicates that they have an identical set of the ribosomal proteins we used. This could often be the case for very closely related populations you want to study. Let’s keep those name in mind and first do a pangenomic analysis.
Pangenomic + Phylogenomics
Here I will not go through the details of pangenomics workflow, but if you are interested in learning more, please read the article “An anvi’o workflow for microbial pangenomics”, and the pangenomic example we used in the Infant Gut Tutorial if you are feeling particularly procrastinative today.
Here is the pangenomic analysis:
# generate anvi'o genomes storage
anvi-gen-genomes-storage -e external-genomes.txt \
-o Salmonella-GENOMES.db
# do the pangenomic analysis --this will take about
# 5 to 10 mins
anvi-pan-genome -g Salmonella-GENOMES.db \
--project-name Salmonella \
--num-threads 8
# display the pangenome
anvi-display-pan -g Salmonella-GENOMES.db \
-p Salmonella/Salmonella-PAN.db
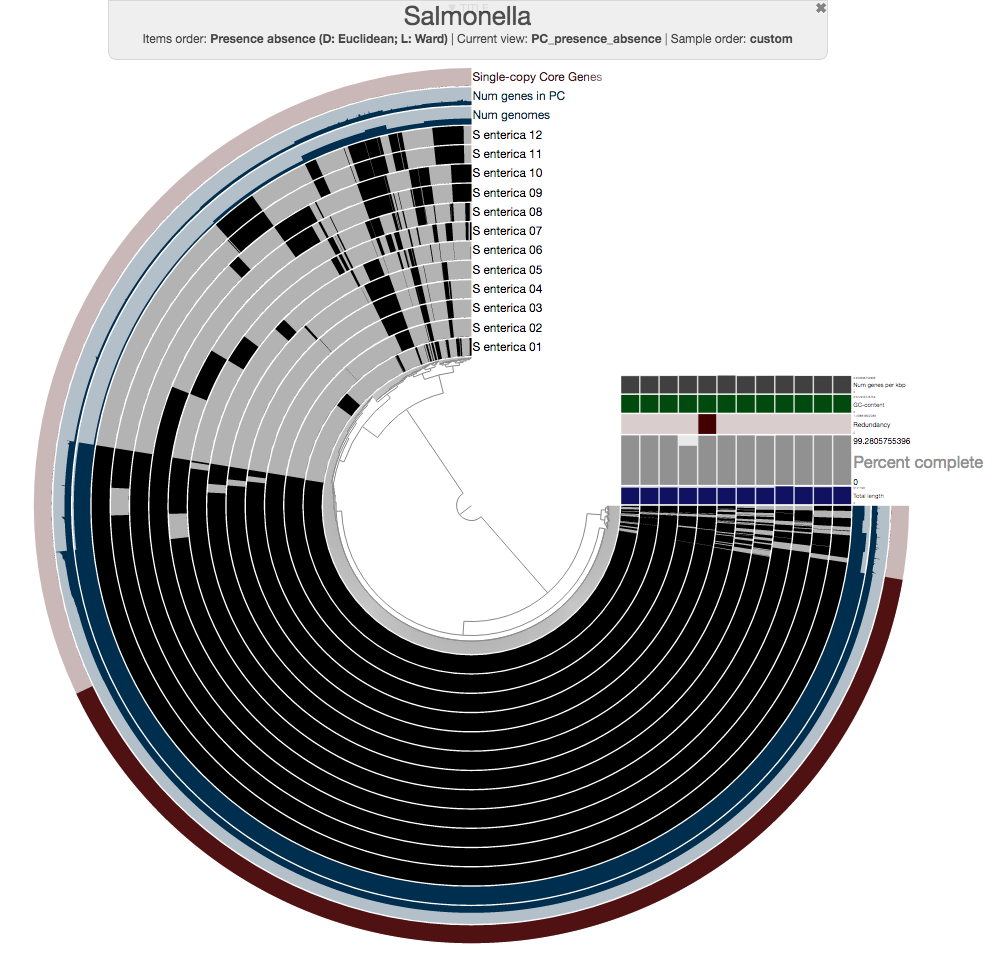
So this is the pangenome of 12 Salmonella strains, and you can already see that those genomes that were flat in the phylogenomic analysis are quite far from being identical.
Expectedly, there is a very large number of core gene clusters that represent genes that occur in every genome (3,456 of them, precisely):
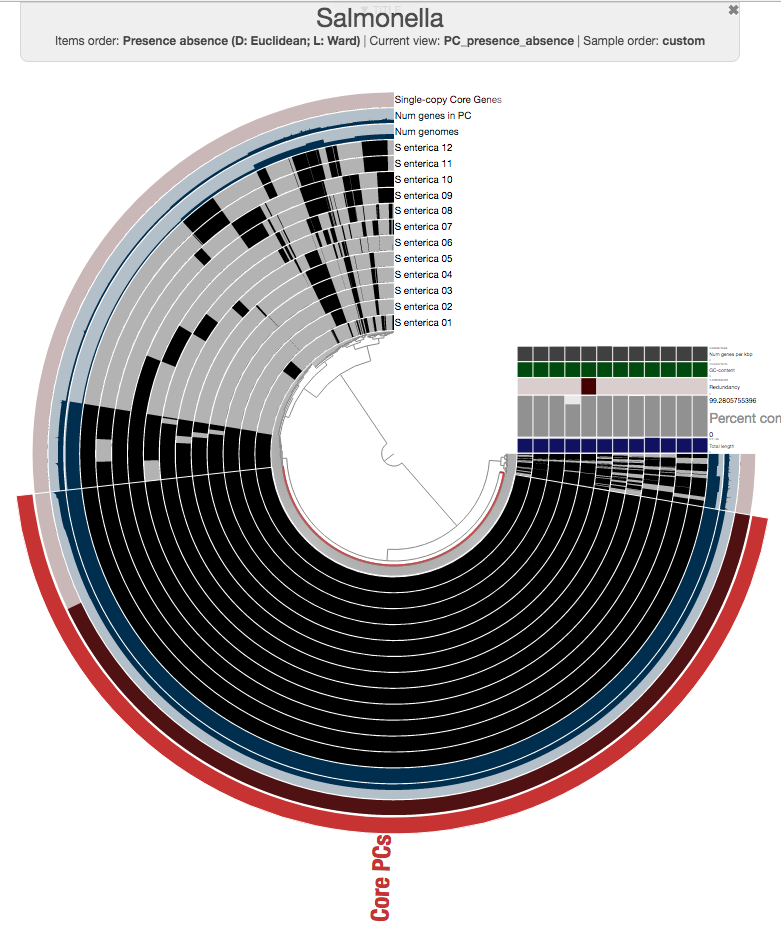
The number of core gene clusters that represent single-copy genes is also quite remarkable, too (there are 3,053 of them)
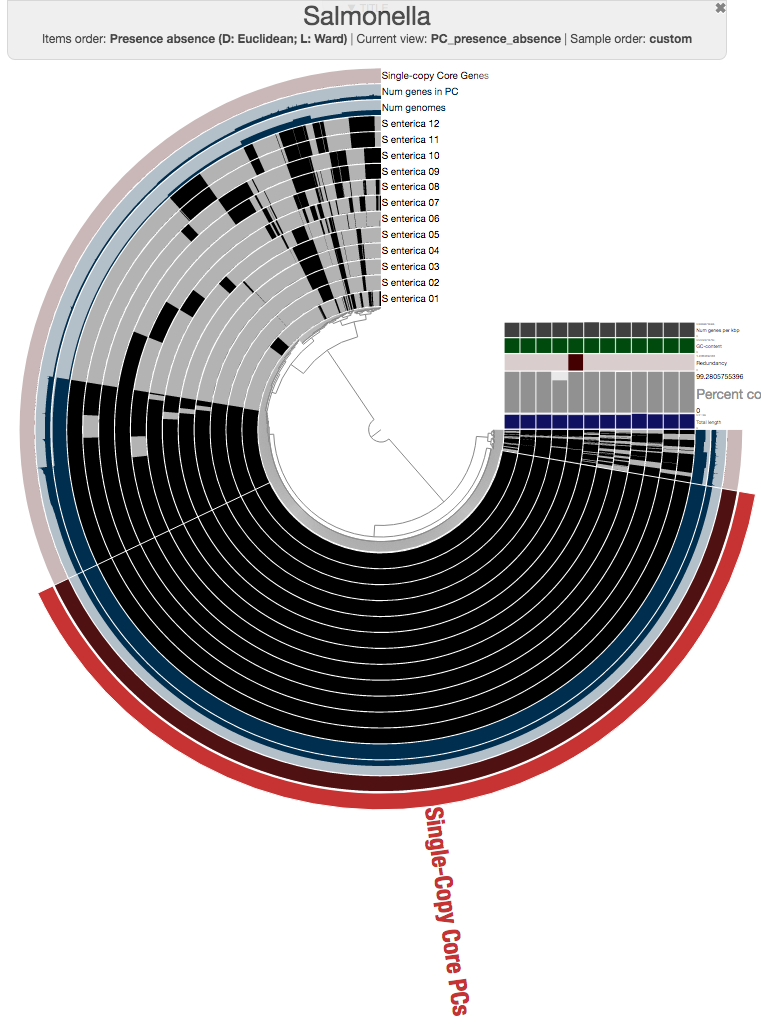
One could certainly store all those genes in a collection and work with them. I will, on the other hand, select 100 genes randomly, and store them in a collection to generate a phylogenomic tree using those:
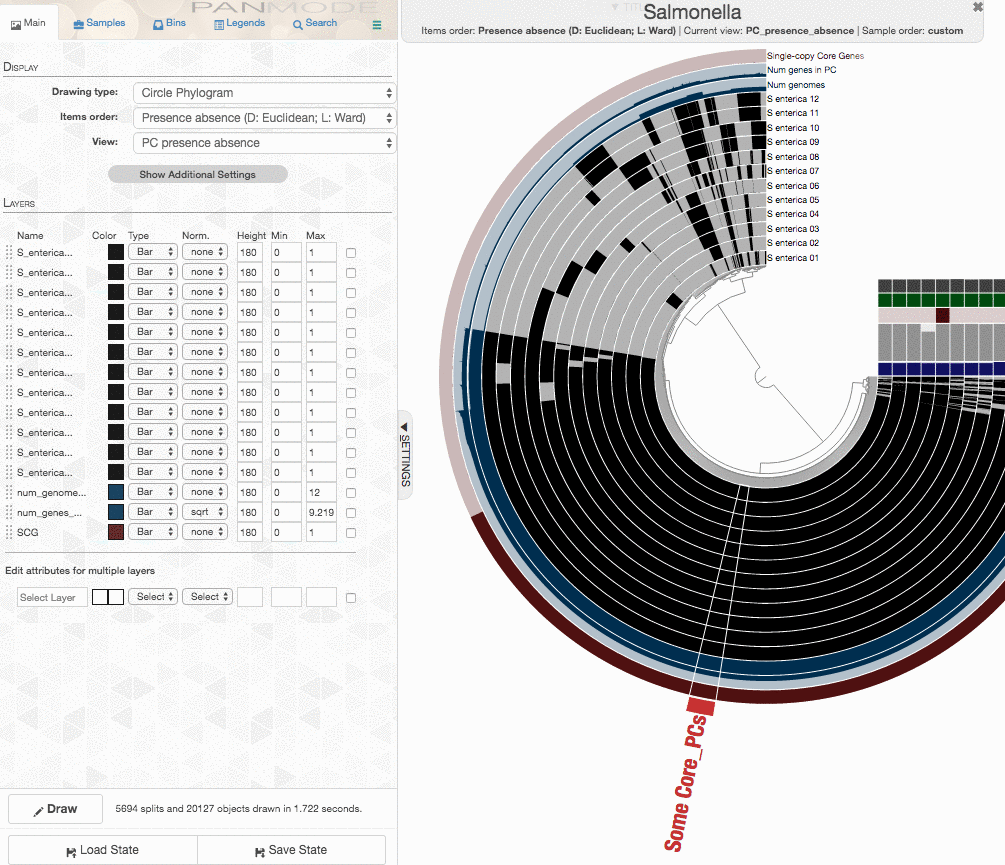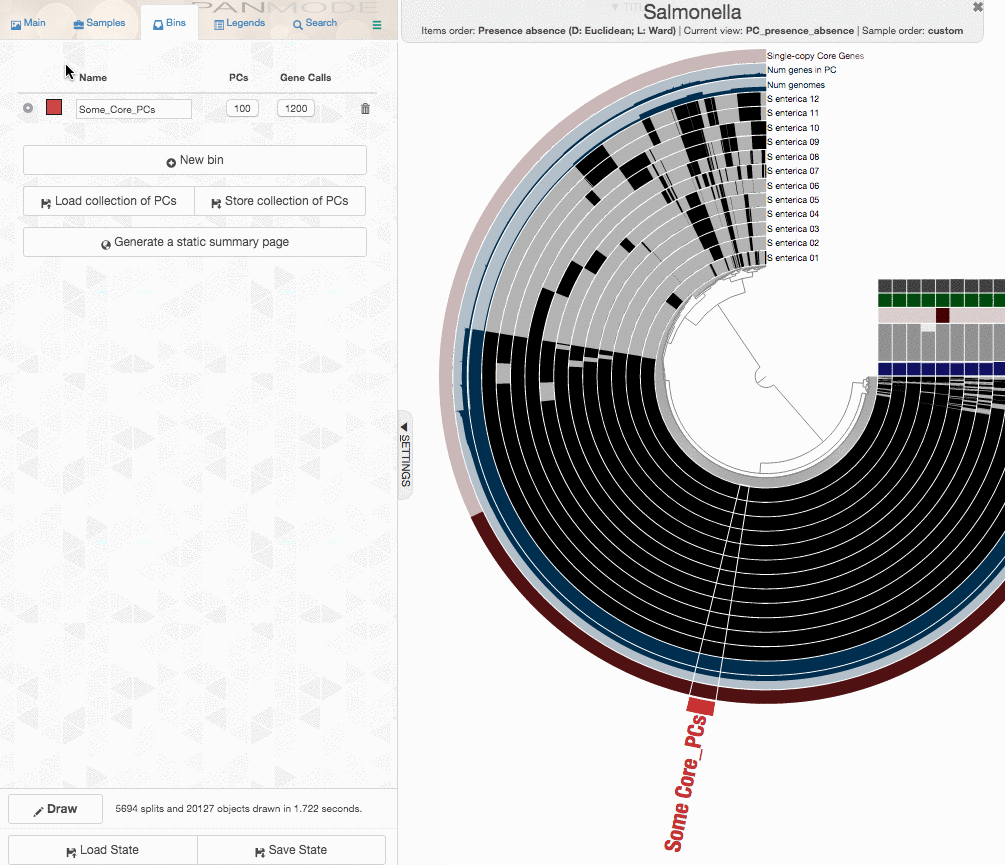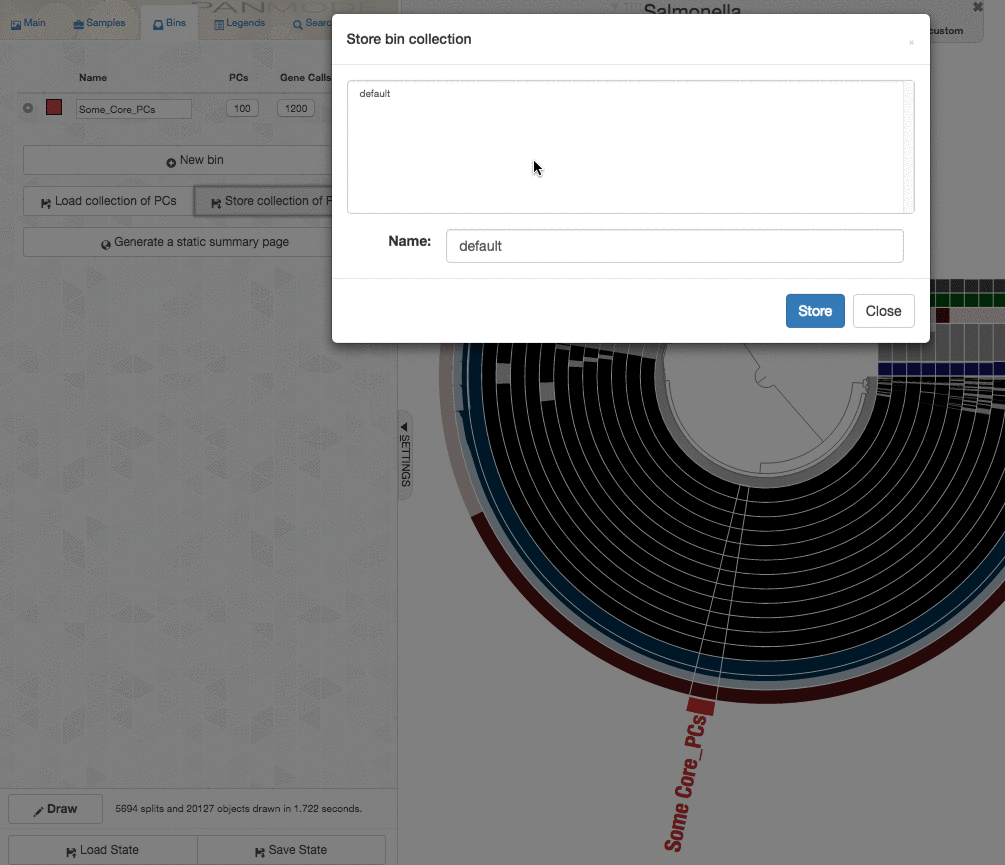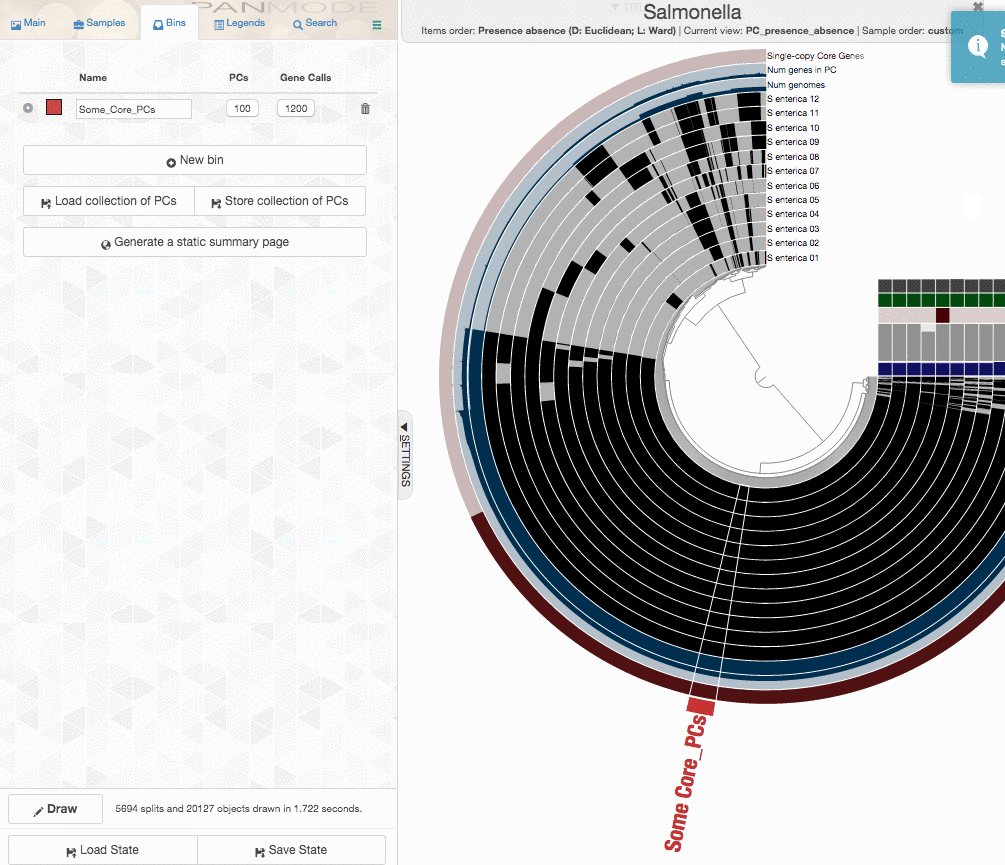
The program anvi-get-sequences-for-gene-clusters is what we will use to export alignments genes in gene clusters. This is also a very capable program, and I urge you to take a look at its help menu and explore other ways to use it for your research. Here, we declare the collection name and the bin id in our anvi’o pan database, and export sequences:
anvi-get-sequences-for-gene-clusters -g Salmonella-GENOMES.db \
-p Salmonella/Salmonella-PAN.db \
--collection-name default \
--bin-id Some_Core_PCs \
--concatenate-gene-clusters \
-o concatenated-proteins.fa
Special Thanks
Until very recently, the programanvi-get-sequences-for-gene-clusters did not have a --concatenate flag either :) We thank Ryan Bartelme for asking for it, and being our beta tester.
Since we now have the FASTA file with concatenated genes, we can generate the phylogenomic tree:
anvi-gen-phylogenomic-tree -f concatenated-proteins.fa \
-o phylogenomic-tree.txt
Good.
If you quickly visualize the resulting tree, you can see that there are no flat branches anymore, indicating that the larger number of core genes we used did convey more signal:
anvi-interactive -p phylogenomic-profile.db \
-t phylogenomic-tree.txt \
--title "Phylogenomics Tutorial Example #4" \
--manual
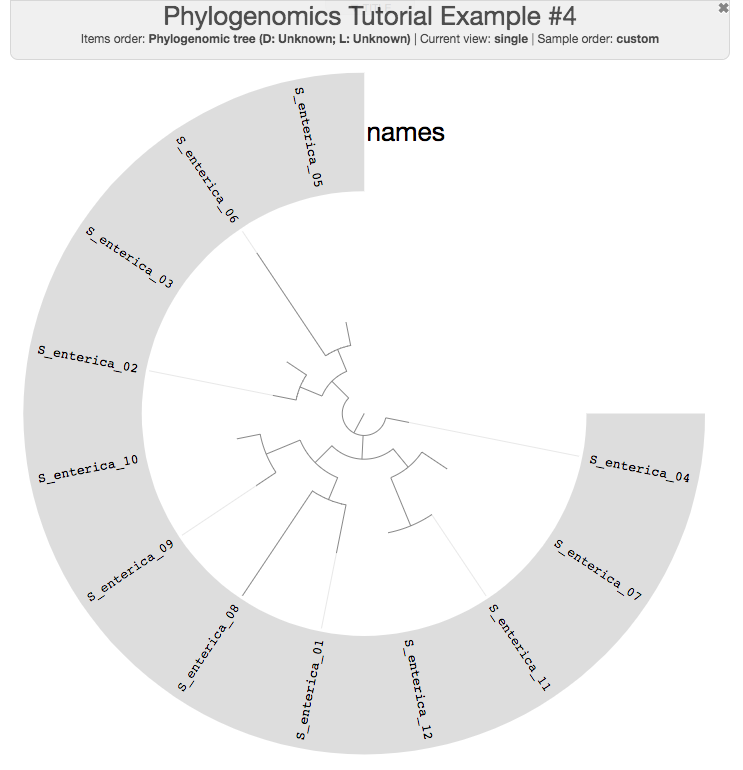
Of course, since I randomly selected 100 core PCs (and maximum-likelihood is not the best way to estimate the relationships between very closely related sequences), there is not much point discussing the biological relevance of this tree.
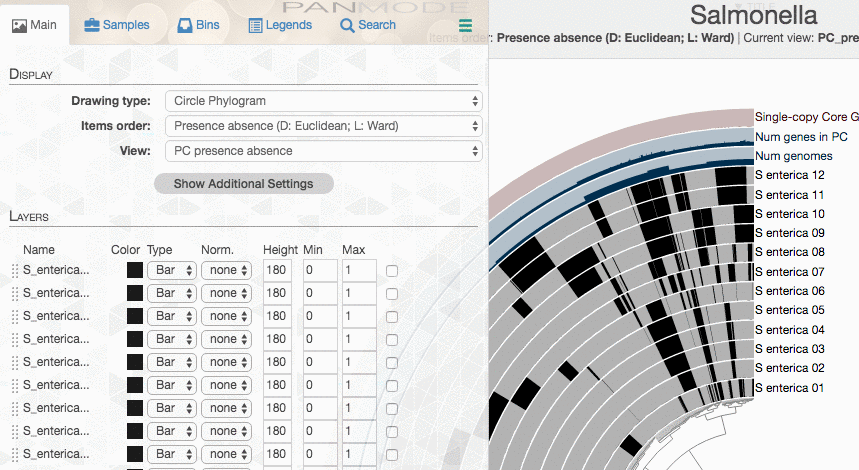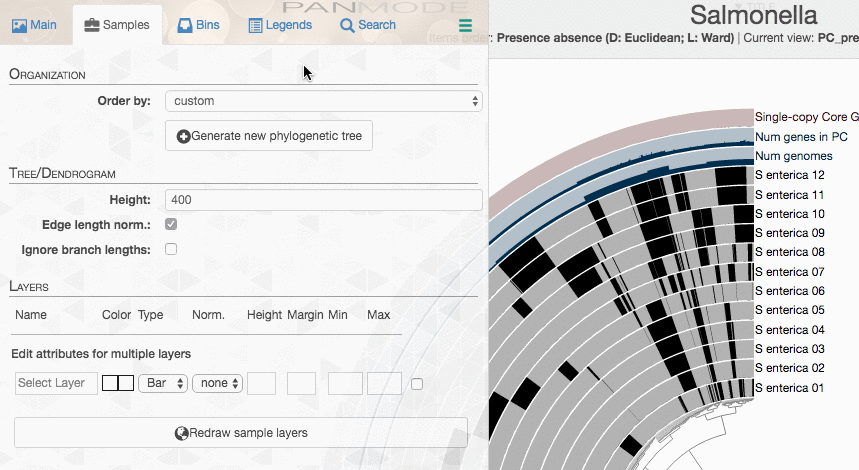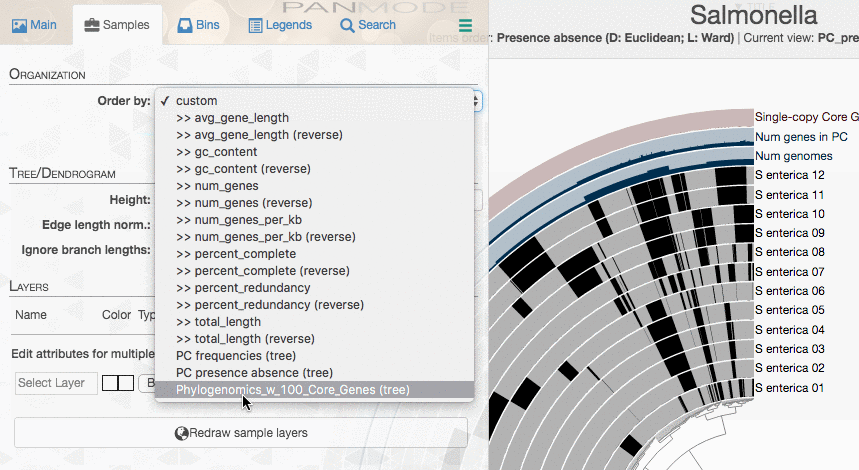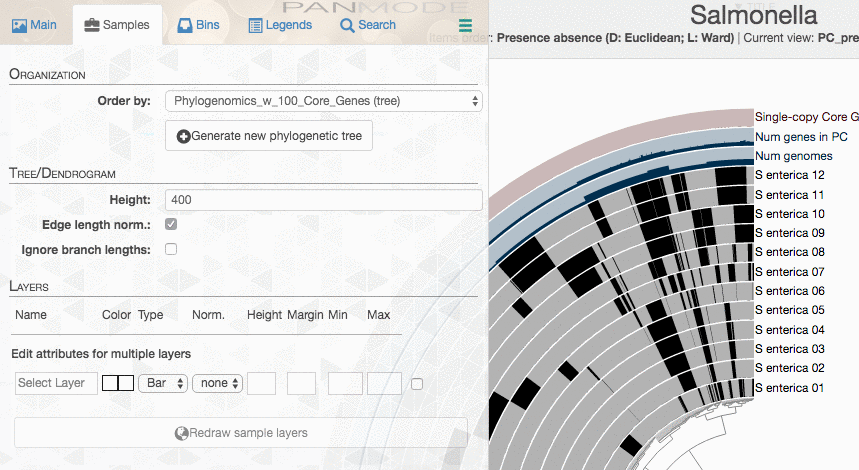
If you want, you can add this organization of your genomes into your pangenome using the additional data subsystem of anvi’o. To add a new genomes order, you will first need to generate a TAB-delimited file like this one as explained here:
| item_name | data_type | data_value |
|---|---|---|
| Phylogenomics_w_100_Core_Genes | newick | ((S_enterica_02:0.0,S_enterica_03:0.0,S_enterica_04:0.0,S_enterica_05:0.0,S_enterica_08:0.0,S_enterica_09:0.0):0.00055,(S_enterica_01:0.00126,S_enterica_07:0.00126)0.453:0.00055,(((S_enterica_11:0.0,S_enterica_12:0.0):0.00126,S_enterica_06:0.00254)0.320:0.00055,S_enterica_10:0.00126)0.000:0.00055); |
Then you can import into your pan database:
anvi-import-misc-data -p Salmonella/Salmonella-PAN.db \
-t layer_orders \
additional-layers-data.txt
Run anvi-display-pan again,
anvi-display-pan -g Salmonella-GENOMES.db \
-p Salmonella/Salmonella-PAN.db
And there you should see your new tree:
Please feel free to ask questions!