
Table of Contents
In this post you will read about the result of an oligotyping analysis that highlights the sexual tranmissibility of Gardnerella vaginalis strains among sexual partners.
This is one of the first stories oligotyping revealed and I hope that it will shed more light on the capacity and limitations of the method. This blog post will communicate mostly the story part of the deal, more than the technical aspect of it. I talk about the software pipeline and how to perform oligotyping on sequencing data in this post.
First, a little bit of context.
When I was a Ph.D. student I was working with Michael J. Ferris in Children’s Hospital, New Orleans. Bacterial vaginosis (BV) was one of the bacterial flora associated medical conditions his lab was interested in. Something that probably is very well known by most of you, which is the fact that clustering and/or classification methods might not be enough to reveal patterns in 16S rRNA ribosomal gene tag sequences, occurred to me for the first time while I was working with the vaginal flora of BV patients.
At this point I need to provide some background information on BV. I’ll be brief (probably to an extent where BV specialists will be upset with me for being so blunt), but if you would like to learn more about the healthy or BV-associated vaginal flora I would suggest you to read this paper from Jacques Ravel et al.: Vaginal microbiome of reproductive-age women.
OK.
Bacterial vaginosis is a very abundant medical condition among women. Besides causing many (mostly personal) inconveniences for patients, it is also linked to preterm birth, which is a huge risk factor for infants to develop short and/or long term health problems. Furthermore, BV patients are more susceptible to sexually transmitted diseases.
BV is manifested by a drastic change in the vaginal flora. While most of the time a healthy woman’s vaginal flora is dominated by species from Lactobacillus genus almost by 100%, all sorts of different, mostly virulent genera in samples collected from BV diagnosed patients can be observed. And one of those virulent bugs that is being observed in symptomatic women is Gardnerella vaginalis (according to some studies it is the most virulent one, and once believed to be the cause of BV).
While I was working with samples that were sequenced from more than 50 women, I realized that Gardnerella vaginalis was very abundant in BV diagnosed women (which was a well known thing). On the other hand, although smaller amounts, Gardnerella vaginalis was also present in normal women (which was another well known thing). I thought, if Gardnerella vaginalis was really a very virulent actor of the disease, there must have been more than one type of it, and what I see in healthy women must be commensal strains. So there was more than one type of Gardnerella vaginalis: through biochemical tests on cultivated Gardnerella vaginalis strains and whole genome studies, people had already shown the existence of various types.
But according to published studies, these differences could not be detected with the 16S rRNA gene amplicons, simply because there was not enough variation at the 16S rRNA level to define different types confidently via 3% OTU clustering or via classification methods that depend on curated databases. So Gardnerella vaginalis was another case of the ineptitude of taxonomy where a name defines many different things with major differences in function, and methods like OTU clustering were not useful to explain their diversity de novo either.
But there was some variation. When I aligned about 70.000 Gardnerella vaginalis sequences from the patients I was working with, I saw it, which eventually convinced me to implement oligotyping.
The variation among all V3V5 sequences that were classified as Gardnerella vaginalis by RDP was much smaller than 3%. Therefore there was one major OTU and a bunch of singletons due to erroneous sequences when they were clustered. So, instead of focusing on sequences as a whole and perform pair-wise comparisons, I wanted to focus only on the subtle but consistent nucleotide variations among these extremely closely related 16S rRNA gene tag sequences.
This is what I did: From all sequences in all datasets, I extracted the ones that were 99+% similar to known Gardnerella vaginalis sequences (which is a very stringent filtering you might say). On these sequences, I performed oligotyping in order to generate oligotyping profiles of Gardnerella vaginalis in every dataset.
The results were very surprising.
Most women presented very distinctive Gardnerella vaginalis oligotype profiles. Actually the remarkable amount of diversity within reads that were identified as Gardnerella vaginalis first concerned me and my colleagues; because we initially thought that there was something wrong with the approach. But we were lucky to have data from the sexual partners of these women. When we included sexual partners’ data (which were collected from penile skin or urethra), results became even more interesting. Here is a figure showing Gardnerella vaginalis diversity of 7 sexual partners:
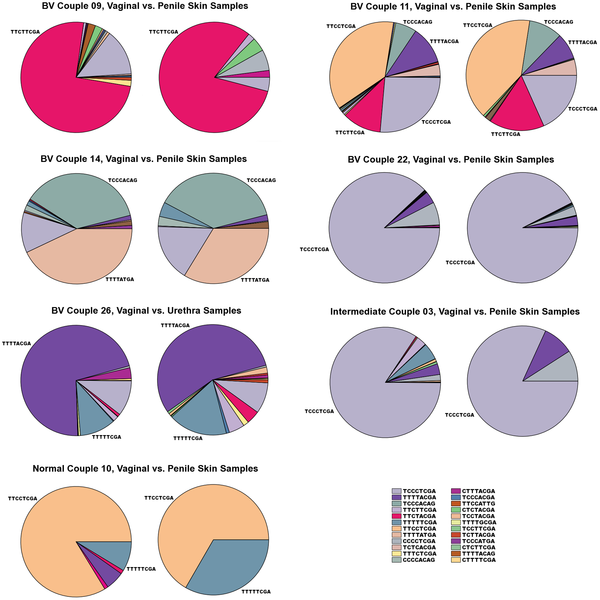
Different colors in the figure correspond to different Gardnerella vaginalis oligotypes. In every pair, the pie chart on the left represents the sample that was collected from the female patient while the other one represents her sexual partners’ penile skin or urethra.
One thing you can immediately see (besides they look almost like fingerprints) is the fact that even though there is great variation among women, different Gardnerella vaginalis types are being maintained in sexual partners. And the maintenance of these similar types is not only at the level of presence-absence, but even their percent abundances are similar.
By just looking at this figure, it could be argued that oligotyping has a biological/ecological significance (since separately PCR-amplified samples present similar profiles among sexual partners), and it can reveal hidden patterns within very closely related taxa that might help researchers understand underlying ecology better (since this diversity you see in this figure has never been shown at this resolution before).
The publication of this story is here, by the way: Exploring the Diversity of Gardnerella vaginalis in the Genitourinary Tract Microbiota of Monogamous Couples Through Subtle Nucleotide Variation.
Of course they didn’t look so clear for every partner in the study. But it is important to remember that the time that partners have been together, condom usage, frequency of sex and being monogamous versus polygamous may have a critical impact on developing similar/distinct looking profiles.
On the other hand I don’t know how resilient these types are (nor do I know their function in the community at this point). But for instance, if either of the partners in one of those couples engages sexual relationships with a new partner, would we see a difference in their Gardnerella vaginalis oligotype profiles? Can some of these types be more associated with the severity or re-occurrence of BV in women? If being exposed to different microbial flora (by means of sexual interaction) changes these profiles, and some of these types are linked to the severity of re-occurrence of the condition, can we say it is more likely to develop BV by having a polygamous relationship?
I don’t know. I am not working specifically on Bacterial Vaginosis.
But at this point if you are interested in such questions, oligotyping might provide some new insights for your own study that other methods can’t.
And by just looking at the figure above, it can be argued that after a sexual relationship, people should remember that there are more to what they carry to the next relationship than just the baggage of emotions!