
Table of Contents
This tutorial was initially written using anvi’o v5.2, but has been updated to include the later addition of anvi-script-process-genbank.
Introduction
When doing a pangenomic analysis with anvi’o, we will often want to integrate some newly recovered genomes with available reference genomes. Currently, it is fairly straightforward as all you need is FASTA files for each genome to call genes and annotate them with functions all in anvi’o. However, this doesn’t take advantage of the existing annotations for publically available reference genomes, and depending on your purposes, you may want this.
It is also possible that you may have already annotated your new isolate or metagenome-assembled genomes (MAGs) with NCBI’s PGAP (Prokaryotic Genome Annotation Pipeline), and may want to use those annotations within anvi’o.
The purpose of this post is to demonstrate a way to incorporate new genomes with reference genomes from NCBI, while maintaining their vetted reference-genome annotations.
The content of this post is a little hacky, but it’s the only way I’ve figured out how to do it so far – plus it’s fun because we get to play with Unix 😃
Data
To make it easier for you to follow this post with your own data, I will go through it by using some of the Prochlorococcus genomes incorporated in the recent Delmont and Eren study.
This study included both isolate and single-cell amplified genomes (SAGs) from Kashtan et al. 2014), and is also used as example data in the stellar pangenomics workflow page (seriously, if you haven’t yet, read through that page sometime if you do or plan to do microbial pangenomics, it’s ever-growing and shows a lot of anvi’o’s pan-functionality). None of these genomes are new genomes in this case, but since isolate genomes are annotated by NCBI’s PGAP when archived in NCBI, and SAGs are not, the SAGs can serve as ‘newly recovered genomes’ to fullfil our purpose here. We will use FASTA files for SAGs, so they will be treated the same way we would treat newly recovered genomes. Here’s the summary of what we’ll be using:
| Genome | Assembly accession | Status | NCBI annotations? | Starting file type |
|---|---|---|---|---|
| MIT9301 | GCA_000015965.1 | Reference isolate genome | Yes | GenBank |
| MIT9314 | GCA_000760035.1 | Reference isolate genome | Yes | GenBank |
| JFNU01 | GCA_000634515.1 | “Newly recovered” genome | No | Fasta |
| JFMG01 | GCA_000635555.1 | “Newly recovered” genome | No | Fasta |
And here’s one way you can download and rename them if you want to follow along:
# creating and entering a new directory
mkdir ~/pan_demo && cd ~/pan_demo
# download each one of them
curl ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/015/965/GCA_000015965.1_ASM1596v1/GCA_000015965.1_ASM1596v1_genomic.gbff.gz -o MIT9301_ref.gbff.gz
curl ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/760/035/GCA_000760035.1_ASM76003v1/GCA_000760035.1_ASM76003v1_genomic.gbff.gz -o MIT9314_ref.gbff.gz
curl ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/634/515/GCA_000634515.1_De_novo_assembly_of_single-cell_genomes/GCA_000634515.1_De_novo_assembly_of_single-cell_genomes_genomic.fna.gz -o JFNU01.fna.gz
curl ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/635/555/GCA_000635555.1_De_novo_assembly_of_single-cell_genomes/GCA_000635555.1_De_novo_assembly_of_single-cell_genomes_genomic.fna.gz -o JFMG01.fna.gz
# decompress all:
gunzip *.gz
The process
As mentioned above, we need to get a little hacky to do this. If you have questions or ways to improve things, please feel free to send a comment down below.
For things to work with the infrastructure currently in place in anvi’o, the same gene-caller needs to be used for all genomes. This is because otherwise the pangenomic workflow will complain about the fact that we are trying to combine genomes whose genes have been identified with different gene-callers. A valid concern on anvi’o’s part, but here we will trick it as we know what we are doing.
To meet this criterion (or more accurately to make anvi’o think we do), I’ve found it easiest to make one contigs database holding all genomes, put them into bins in a collection, and then send them into anvi-pan-genome as “internal genomes”.
Here’s how we’ll do this throughout this post:
-
Combine all reference GenBank files together and parse for anvi’o
-
Call genes for the newly recovered genomes and combine them with the reference files
-
Generate the “master” contigs database and import functions
-
Add genome/bin info
-
Create the internal genomes file
-
Pangenomification
Combining GenBank files and parsing for anvi’o
Here we are going to combine the reference GenBank files (that hold annotations and sequences) together:
cat *_ref.gbff > all_refs.gbff
And now we need to parse this GenBank file into the needed files for generating our contigs database.
We can use the program anvi-script-process-genbank within anvi’o to do this. Because anvi’o expects the gene-calling annotation source of all genomes being incorporated into a pangenome analysis to be the same, we are going to specify the --annotation-source as “prodigal”, even though the NCBI PGAP annotations were done a little more extensively:
anvi-script-process-genbank -i all_refs.gbff \
--output-gene-calls all_refs_gene_calls.tsv \
--output-functions all_refs_functions.tsv \
--output-fasta all_refs.fa \
--annotation-source prodigal
Side Note
If you are working with NCBI genbank files that are not yet publicly available, you may get an error from anvi-script-process-genbank due to the genbank file not being in the “proper” format that is expected by the underlying biopython module that handles parsing it. I have a small program in my bioinformatics tools package to get around this. If needed, we can install that with conda like so (it’d be best to put it in its own environment, not within the anvi’o environment):
conda create -y -n bit -c conda-forge -c bioconda -c defaults -c astrobiomike bit
conda activate bit
Then we can clean up the format with the following command (renaming it afterwards to match the code above):
bit-genbank-locus-clean-slate -i all_refs.gbff -o all_refs.gbff-clean
mv all_refs.gbff-clean all_refs.gbff
And after switching back out of the bit environment into the proper anvi’o environment, we’d be ready to use the anvi-script-process-genbank command above 🙂
Now back to our regularly scheduled post
anvi-script-process-genbank outputs 3 files: 1) an external gene calls file, all_refs_gene_calls.tsv; 2) a FASTA file, all_refs.fa; and 3) a functional annotation file, all_refs_functions.tsv. Files 1 and 2 are needed for creating our contigs database, and file 3 right after that to import our functional annotations. But right now all 3 of these just hold the reference genomes. That’s fine for the annotations file (3) at this point because the reference genomes have no annotations yet. But we need to add gene calls (to file 1) and sequences (to file 2) for our new genomes in order to be able to create our contigs database with all.
Calling genes for the newly recovered genomes and combining with reference files
First we’re going to clean the headers of our new genome FASTA files with anvi-script-reformat-fasta, and explicitly add the genome identifier. This will help us with creating our collection later.
Then we’re going to call genes on our new genomes so we can add them to the all_refs_gene_calls.tsv file we just created. We will do this with prodigal (v2.6.3 here) on our own, and then add them ourselves.
We’ll run these here in a loop – if you are new to a Unix-like command line and/or loops and want to learn their awesome power, check out this page here :)
for genome in $(ls *.fna | cut -f1 -d ".")
do
anvi-script-reformat-fasta -o "$genome"_clean.fa \
--prefix "$genome" \
--simplify-names \
"$genome".fna
prodigal -f gff \
-c \
-i "$genome"_clean.fa \
-o "$genome".gff
done
Now we are going to concatenate the “cleaned” FASTA files with our references:
cat all_refs.fa *_clean.fa > all_genomes.fa
Here is where the command-line stuff is going to start getting a little heavy. If you aren’t comfortable with it yet, and want to get better acquainted (which is infinitely worthwhile if you are going to be doing this stuff at all regularly), a great place to start is here :)
Here we are going to parse the gff files from prodigal to match the formatting for the anvi’o external gene calls file. This is a tab-delimited file (with a header) with 6 columns: “gene_callers_id”, “contig”, “start”, “stop”, “direction”, “partial”, “source”, and “version”. Here is one way to build those last 5 columns:
# getting all gene-call lines only
grep -hv "#" *.gff > all_new_genomes.gff.tmp
# building columns for external gene calls file
cut -f1 all_new_genomes.gff.tmp > contig.tmp
# subtracting 1 to match the 0-based counting used in
# anvi'o (it's closed end, so don't need to modify stops)
cut -f4 all_new_genomes.gff.tmp | awk '{print $1-1}' > start.tmp
cut -f5 all_new_genomes.gff.tmp > stop.tmp
cut -f7 all_new_genomes.gff.tmp > initial_direction.tmp
# determining the direction of the gene call
for i in $(cat initial_direction.tmp)
do
if [ $i == + ]
then
echo "f"
else
echo "r"
fi
done > direction.tmp
# because we specified the `-c` option when running prodigal,
# we have no partial genes
for i in $(cat direction.tmp); do echo "0"; done > partial.tmp
for i in $(cat direction.tmp); do echo "prodigal"; done > source.tmp
for i in $(cat direction.tmp); do echo "v2.6.3"; done > version.tmp
Now we need that first column with unique gene_callers_id numbers for the new gene calls we’re adding. To get that, we want to know what is the maximum number for gene calls in the current gene calls file, and go from there by incrementing that number per new gene call. Here’s one way to do that:
# this counts the header, but that's ok because we want to
# start 1 higher than already exists anyway :)
start_count=$(wc -l all_refs_gene_calls.tsv | awk '{print $1}')
# no header in this file
new_genes=$(wc -l direction.tmp | awk '{print $1}')
# getting final gene call position
stop_count=$(echo "$start_count + $new_genes" -1 | bc)
# spanning range to create new gene caller ids
for i in $(seq $start_count $stop_count)
do
echo $i
done > gene_callers_id.tmp
NOW we can put together all these temporary files into a single one:
# sticking all together
paste gene_callers_id.tmp \
contig.tmp \
start.tmp \
stop.tmp \
direction.tmp \
partial.tmp \
source.tmp \
version.tmp > adding_gene_calls.tmp
# combining with the rest
cat all_refs_gene_calls.tsv \
adding_gene_calls.tmp > all_genomes_gene_calls.tsv
# deleting intermediate files
rm *.tmp
Creating the “master” contigs database
Now we have the files needed to create our “master” contigs database that holds all of the genomes we want to pangenomify. In this case, since we are not looking at coverage or doing any binning, I am skipping splitting of contigs to save time by providing -L -1:
anvi-gen-contigs-database -f all_genomes.fa \
-o contigs.db \
-n Pro_genomes \
--external-gene-calls \
all_genomes_gene_calls.tsv \
--split-length -1
And importing our functional annotations (for the reference genomes, there are none for our “new” ones in here):
anvi-import-functions -c contigs.db \
-i all_refs_functions.tsv
At this point we can also scan for single-copy genes with anvi-run-hmms and annotate all genomes with anvi-run-ncbi-cogs and/or anvi-run-pfams (though I’m skipping anvi-run-pfams here for time as well):
# run default anvi'o HMMs
anvi-run-hmms -c contigs.db \
--num-threads 4
# run NCBI COGs
anvi-run-ncbi-cogs -c contigs.db \
--num-threads 4
Adding genome/bin info
Now all our genomes are in a single contigs database. To access to each genome separately, we need to import a collection, which is a 2-column, tab-delimited (headerless) file holding inormation regarding which contig belongs to which bin (or genome in our case).
This requires being able to identify your genomes based on their contig identifiers as we’re going to do here, if you have trouble with this with your own data, feel free to message me and I will try to help out :)
Here’s one way to make that file with the current situation:
# looping through the "non-reference" genome fasta files:
for genome in $(ls *_clean.fa | sed 's/_clean.fa//')
do
grep ">" "$genome"_clean.fa | cut -f1 -d " " | tr -d ">" > "$genome"_contigs.tmp
for contig in $(cat "$genome"_contigs.tmp)
do
echo "$genome"
done > "$genome"_name.tmp
paste "$genome"_contigs.tmp "$genome"_name.tmp > "$genome"_for_cat.tmp
done
# looping through the reference genomes' genbank files:
for genome in $(ls *_ref.gbff | cut -f1 -d "_")
do
grep "LOCUS" "$genome"_ref.gbff | tr -s " " "\t" | cut -f2 > "$genome"_contigs.tmp
for contig in $(cat "$genome"_contigs.tmp)
do
echo "$genome"
done > "$genome"_name.tmp
paste "$genome"_contigs.tmp "$genome"_name.tmp > "$genome"_for_cat.tmp
done
# catting all together and removing intermediate files:
cat *_for_cat.tmp > collection.tsv && rm *.tmp
# and for a sanity check we can make sure our file is the right size:
grep -c ">" all_genomes.fa
# 631
wc -l collection.tsv
# 631 collection.tsv
# and check out our genome/bin names:
cut -f2 collection.tsv | uniq -c
# 377 JFMG01
# 237 JFNU01
# 1 MIT9301
# 16 MIT9314
Now importing our collection:
# we will need a blank profile to store our collection
anvi-profile -c contigs.db \
-o profile \
-S profile \
--blank-profile \
--min-contig-length 0 \
--skip-hierarchical-clustering
# now we can import our collection
anvi-import-collection collection.tsv \
-c contigs.db \
-p profile/PROFILE.db \
-C Pro_genomes \
--contigs-mode
Creating the internal genomes file
This file is needed to set things up for the pangenomic analysis. The format of this file is documented here, and for internal genomes as we’re using here, it is tab-delimited with 5 columns with the headers “name”, “bin_id”, “collection_id”, “profile_db_path”, and “contigs_db_path”
Here is how I put this one together:
echo -e "name\tbin_id\tcollection_id\tprofile_db_path\tcontigs_db_path" > header.tmp
cut -f2 collection.tsv | uniq > name_and_bin_id.tmp
for i in $(cat name_and_bin_id.tmp); do echo "Pro_genomes"; done > collection_id.tmp
for i in $(cat name_and_bin_id.tmp); do echo "$PWD/profile/PROFILE.db"; done > profile_db_path.tmp
for i in $(cat name_and_bin_id.tmp); do echo "$PWD/contigs.db"; done > contigs_db_path.tmp
paste name_and_bin_id.tmp name_and_bin_id.tmp collection_id.tmp profile_db_path.tmp contigs_db_path.tmp > body.tmp
cat header.tmp body.tmp > internal_genomes.tsv && rm *.tmp
And now we’re all set :)
Pangenomification
Just to wrap things up with a nice bow, here is finally running the pangenome analysis:
anvi-gen-genomes-storage -i internal_genomes.tsv \
-o Prochlorococcus-GENOMES.db
anvi-pan-genome -g Prochlorococcus-GENOMES.db \
-n Prochlorococcus-PAN \
--num-threads 4
Now NCBI’s PGAP annotations are a part of our pangenome. This means, for example, if we are interested in an iron-sulfur ATP-binding protein, “ChlL”, that we know to be in our reference genome MIT9314 under its protein accession “KGG03723.1”, we can search for that accession in our pangenome and see if our new genomes have genes in that cluster:
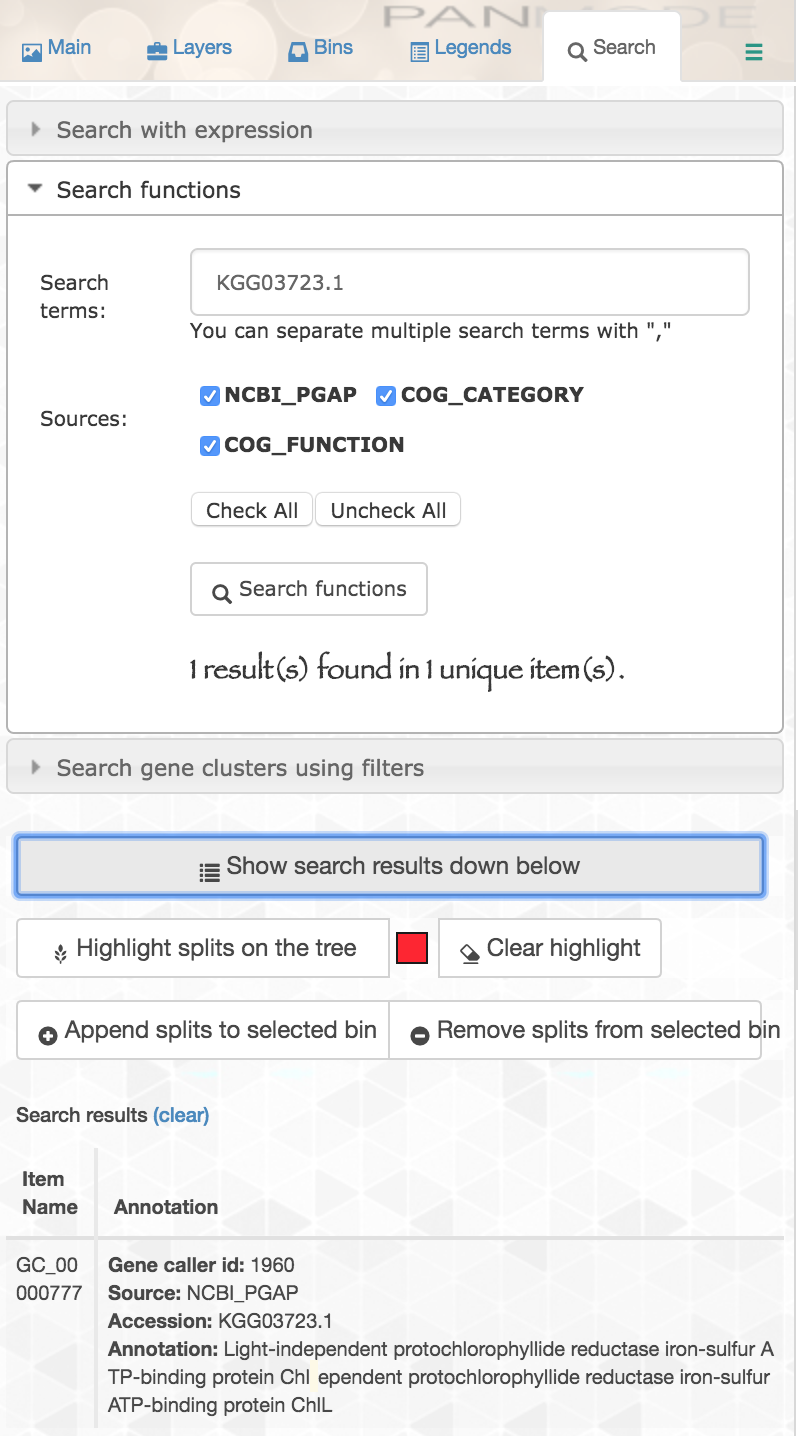
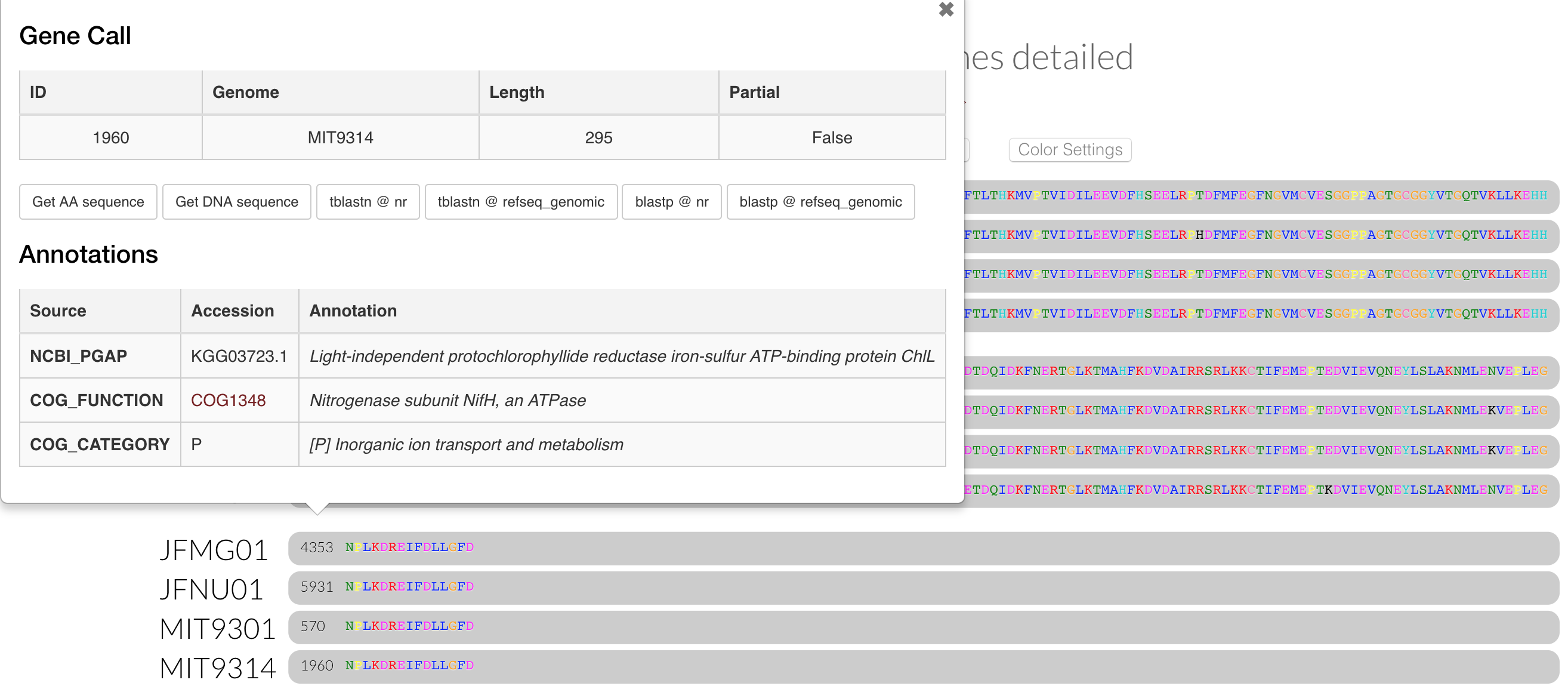
If we inspect that cluster, we can see the two new genomes do possess genes in it:
And of course when we summarize the pangenome with anvi-summarize, these annotations are present in there as well, giving us more room for parsing gene clusters as we’d like.
Well that’s pretty much it… for getting the processing done anyway…
But as we know, this is actually just the beginning. Again, do look over the pangenomic workflow page if you haven’t yet, or haven’t in a while. The anvi’o team keeps adding more and more awesome functionality – like the new homogeneity indices for gene clusters, for instance.