

Table of Contents
- Who is this post for?
- Introduction
- Breaking it down into (6) components
- (1) and (2): Storing copies of the InteracDome datasets and the corresponding HMM profiles
- (3) Running the hidden Markov model (HMM)
- (4) Parsing HMMER’s standard output file
- (5) Filtering HMM hits
- (5) Matching binding frequencies to the genes’ residues
- (6) Storing the per-residue binding frequencies into the contigs database
- Conclusion
This feature is implemented in anvi’o v7 and later. You can identify which version you have on your computer by typing anvi-self-test --version in your terminal.
The latest version of anvi’o is v7.1. See the release notes.
Who is this post for?
NOTE: For a more practical tutorial on the same topic, please visit the infant gut tutorial
This post is like a technical diary for my implementation of InteracDome into anvi’o. The primary purpose of this is to provide all the technical details in one place, so that (1) people who inevitably use this in anvi’o know what is going on under the hood, and (2) so people digging into the codebase for debugging or extending features understand why I made certain decisons. It is thus a very technical blog post, and in that sense is quite distinct from other stuff we write about. But if you think you may be interested, please keep reading. I will not pretend that this has a start middle and end–it is more a reference for all the decisions I made. Just keep in mind that I will not be shying away from sharing code snippets from the codebase, so if you’re code-shy, this is your trigger warning.
This post is not version-controlled. The codebase is dynamic and will inevitably change, but this post will (probably) not, and is therefore merely a snapshot of what once was (July 20th, 2020). I include it in the hopes it provides conceptual clarity, and hope it is not taken too seriously, or an accurate representation of anvi’o’s codebase.
Introduction
Far too often we do not know what our SNV, SAAV, and SCV data mean. The problem is that our data output is nucleotide sequence, which defines the blueprint for function, but is a very abstracted output from what we are trying to learn about. As eloquently put by Harms and Thornton, as a practical convenience, we as evolutionary biologists have a tendency to “treat molecular sequences as mere strings of letters, the patterns of which carry the traces of historical processes, rather than as functioning objects for which the physical properties determine their behaviour”. It is from these patterns that we can learn so much by analyzing variant data derived from metagenomes. However, given that the SNV/SAAV/SCV (I’ll just call them variants from now on) patterns by themselves offer zero insight into fitness, within our field, variant data is most commonly used to identify and track “strains”. And to be honest, that saddens me because there is so much more potential than using them as ecological markers.
To bridge a gap between metagenomic sequence variants and the structural biology of gene products that underpin function and fitness is no easy task, but I think its critical if we want empower our findings with biochemical information. (As a small step in this direction, Özcan, Meren, and I developed anvi’o structure, a way to visualize metagenomic sequence variants directly on predicted protein structures).
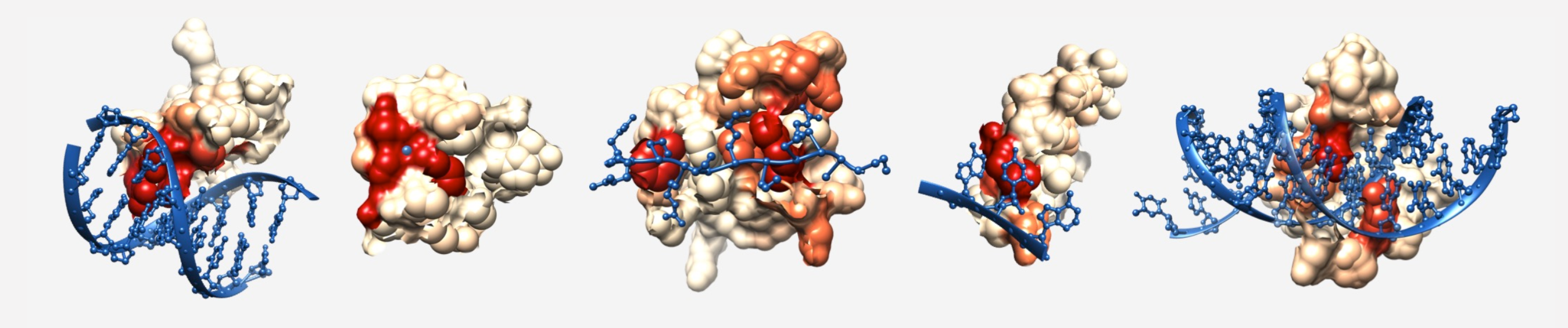
Related to this concept, one of the most transformative talks I have ever attended was given by Dr. Mona Singh about this paper (Kobren and Singh, 2018). Kobren and Singh took every Pfam family and searched for whether or not any members of the family had crystalized structures that co-complexed with bound ligands. If so, then they calculated 3D distance scores of each residue to the ligand(s) and used these distances as an inverse proxy for binding likelihood (the more likely you are to be involved in the binding of a ligand, the closer you are in physical space to the ligand). They aggregated the totality of these results and ended up with thousands of Pfam IDs for which they could attribute per-residue ligand-binding scores to the Pfam’s associated hidden Markov model (HMM) profile. They used this to extend our knowledge of the human proteome, however when I was listening to the talk all I was thinking about was applying it to metagenomics.
They entitled their software, InteracDome, and there is an online server where you can give an amino acid sequence, and their server will run an HMM of your sequence against the InteracDome database and attribute estimated binding scores for each residue in your sequence. That is basically exactly what I wanted to do, except I wanted to extend this to the potentially massive number of genes that can typically found in anvi’o contigs databases. Furthermore, I wanted to store all the information in the contigs database for further data analysis and visualization, as is the anvi’o way.
Breaking it down into (6) components
Here are the bird’s-eye-view components of InteracDome’s implementation into anvi’o:
- Storing a local copy of InteracDome’s tab-separated files
- Storing the HMM profiles that the contig database’s genes are searched against
- Running the HMM
- Parsing HMMER’s standard output file
- Filtering HMM hits
- Matching binding frequencies to the gene’s residues
- Storing the per-residue binding frequencies into the contigs database
(1) and (2): Storing copies of the InteracDome datasets and the corresponding HMM profiles
Description of tab-separated InteracDome files
What are these tab-separated files, you say? Well they contain the binding frequencies associated to each match-state (read as: residue) of the HMM. Here is what one looks like (be sure to scroll right to get a complete picture of the table):
| pfam_id | domain_length | ligand_type | num_nonidentical_instances | num_structures | binding_frequencies |
|---|---|---|---|---|---|
| PF00001_7tm_1 | 268 | 1WV | 3 | 4 | 0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.460539215686,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.539460784314,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.360294117647,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0 |
| PF00001_7tm_1 | 268 | 2CV | 7 | 10 | 0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.103823009901,0,0,0.103823009901,0,0,0,0,0,0.207401960435,0,0,0,0.155734514852,0,0,0.577037641019,0,0,0.572218731277,0.0519115049506,0,0.310492792233,0.473458690485,0,0,0,0,0,0.155490455484,0,0,0,0,0,0.109873556499,0,0,0.051667445583,0,0.0519115049506,0,0,0,0.574814741526,0.371783032133,0,0,0,0,0,0,0,0,0,0,0,0,0,0.582709105685,0.681678668828,0.112286030157,0,0,0,0,0,0,0,0.0517419453701,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.0519115049506,0.207646019802,0,0,0,0,0,0,0.0519115049506,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.0519115049506,0,0.103823009901,0.155734514852,0,0,0.0519115049506,0.0519115049506,0,0,0,0,0,0,0,0,0,0,0.0519115049506,0,0.051667445583,0,0,0,0.0544330154125,0,0,0,0,0,0.051667445583,0,0,0,0,0,0,0 |
| PF00001_7tm_1 | 268 | ERM | 6 | 5 | 0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1.0,0.123851117406,0,0,1.0,1.0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.123851117406,0.258781869688,0.134930752282,0.125645262827,0.125645262827,0,0,0.490352533837,0.296797293044,0,0.125220333648,0.134930752282,0,0,0,0,0,0,0.134930752282,0,0.31920050362,0,0,0.296797293044,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.296797293044,0,0,0.490352533837,0,0,0,0.490352533837,0.123851117406,0.249071451054,0.318775574441,0.193555240793,0,0.125220333648,0,0.249496380233,0,0,0,0.125645262827,0,0,0,0,0.431728045326,0.296797293044,0,0.123851117406,0.422017626692,0,0,0.249071451054,0,0,0.123851117406,0.125220333648,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0 |
| PF00003_7tm_3 | 238 | SM_ | 3 | 4 | 0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.248912097476,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.248912097476,0,0,1.0,1.0,0,0,0.498259355962,0.751087902524,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.248912097476,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.249347258486,0.249347258486,0.497824194952,0,0.249347258486,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.249347258486,0,0.497824194952,0,0.249347258486 |
| PF00003_7tm_3 | 238 | ALL_ | 3 | 4 | 0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.248912097476,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.248912097476,0,0,1.0,1.0,0,0,0.498259355962,0.751087902524,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.248912097476,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.249347258486,0.249347258486,0.497824194952,0,0.249347258486,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.249347258486,0,0.497824194952,0,0.249347258486 |
| PF00004_AAA | 132 | SM_ | 205 | 166 | 0.0114604627231,0.00477140578191,0.00159046859397,0.00573253589269,0.00636187437588,0.0156074891605,0.151915630049,0.00636187437588,0.165530516241,0.00465063703568,0.7665876828,0.782039254129,0.638852101367,0.0148199070736,0.0197556615281,0.0659386849446,0.0233671491129,0.0274992668572,0.0068758651402,0.0243892439834,0.016548822645,0.0175596896304,0.0235691023598,0.0148281476804,0.0108389692647,0.0200299870214,0.0158208194559,0.136698750907,0.0183922951312,0.0108666290365,0.00319897110896,0.00352559062884,0.00340017637127,0.00310366737855,0.00743451388943,0,0,0.00226506315454,0.00226506315454,0.00272300017124,0,0.00159046859397,0.00435666305088,0.00833301017716,0.00674254158319,0.00318093718794,0.00871332610176,0.00871332610176,0.00594713164485,0.00636187437588,0.00477140578191,0.00636187437588,0.00159046859397,0.00159046859397,0,0,0,0,0.0166834633166,0.0124614217996,0.0123052272426,0.00332776801496,0,0,0.0235604250871,0.0205062852754,0.00159046859397,0.00159046859397,0.00443759986205,0.0108022602052,0,0.00758910697789,0.042872742652,0,0,0,0.0205374756636,0,0,0,0.00346838508927,0.00216787556951,0.00390892326059,0.00781784652118,0.00604666604827,0.00768955958345,0.0109017146314,0.012587618788,0.00954249832977,0.0143484218372,0.00116949287805,0.0021871566269,0,0.0243284702568,0.00283095932639,0.00102104179321,0.00897890971083,0.00204208358642,0.0122165343967,0,0,0,0,0.00332776801496,0.00649934147293,0.014056198686,0.00317157345797,0,0.00410153185904,0.0647003263302,0.00826993895804,0.00378063632286,0,0.0446638329233,0.000961349923678,0.00186720282586,0.00186260580663,0.0444138756133,0.00340073494863,0.00628002363626,0.0196825372299,0.00198595471452,0.0189300031233,0.0107861573734,0.00416646190614,0.00314850655058,0.0220221369003,0.00639178475723,0.0043743132538,0.00886361588898,0.0110906488313,0.0021871566269 |
| PF00004_AAA | 132 | DRUGLIKE_ | 205 | 166 | 0.0114604627231,0.00477140578191,0.00159046859397,0.00573253589269,0.00636187437588,0.0156074891605,0.151915630049,0.00636187437588,0.165530516241,0.00465063703568,0.7665876828,0.782039254129,0.638852101367,0.0148199070736,0.0197556615281,0.0659386849446,0.0233671491129,0.0274992668572,0.0068758651402,0.0243892439834,0.016548822645,0.0165092215714,0.0235691023598,0.0137776796214,0.0108389692647,0.0200299870214,0.0158208194559,0.136698750907,0.0183922951312,0.0108666290365,0.00319897110896,0.00352559062884,0.00340017637127,0.00310366737855,0.00743451388943,0,0,0.00226506315454,0.00226506315454,0.00272300017124,0,0.00159046859397,0.00435666305088,0.00833301017716,0.00674254158319,0.00318093718794,0.00871332610176,0.00871332610176,0.00594713164485,0.00636187437588,0.00477140578191,0.00636187437588,0.00159046859397,0.00159046859397,0,0,0,0,0.0156329952576,0.0124614217996,0.0123052272426,0.00332776801496,0,0,0.0235604250871,0.0205062852754,0.00159046859397,0.00159046859397,0.00443759986205,0.0108022602052,0,0.00758910697789,0.042872742652,0,0,0,0.0205374756636,0,0,0,0.00346838508927,0.00216787556951,0.00390892326059,0.00781784652118,0.00604666604827,0.00768955958345,0.0109017146314,0.012587618788,0.00954249832977,0.0143484218372,0.00116949287805,0.0021871566269,0,0.0243284702568,0.00283095932639,0.00102104179321,0.00897890971083,0.00204208358642,0.0111660663377,0,0,0,0,0.00332776801496,0.00649934147293,0.014056198686,0.00317157345797,0,0.00410153185904,0.0647003263302,0.00826993895804,0.00378063632286,0,0.0446638329233,0.000961349923678,0.00186720282586,0.00186260580663,0.0444138756133,0.00340073494863,0.00628002363626,0.0196825372299,0.00198595471452,0.0189300031233,0.0107861573734,0.00416646190614,0.00314850655058,0.0220221369003,0.00639178475723,0.0043743132538,0.00886361588898,0.0110906488313,0.0021871566269 |
| PF00004_AAA | 132 | ANP | 12 | 16 | 0,0,0,0,0,0,0.293054223957,0,0.220890723765,0,0.902585657643,0.902585657643,0.902585657643,0,0.0365564138796,0.149544866261,0.0365564138796,0,0,0,0,0,0,0,0,0,0,0.11424794602,0,0,0,0,0.0359910406715,0.0608579284774,0,0,0,0,0,0,0,0,0,0.0608579284774,0.0608579284774,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.0419485401186,0,0,0,0,0,0,0,0.14472176965,0,0,0,0.18127818353,0,0,0,0.0365564138796,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.11424794602,0,0.11424794602,0,0,0,0,0,0,0,0,0,0,0.129010569799,0,0,0,0.112988452381,0,0,0,0.112988452381,0,0,0,0,0,0.0419485401186,0,0,0,0,0,0,0,0 |
| PF00004_AAA | 132 | PEPTIDE_ | 6 | 9 | 0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.12131242741,0.653927119629,0.188346883469,0,0,0,0,0,0,0,0,0,0,0,0,0,0.251899438637,0.0941734417344,0.0941734417344,0,0,0.251899438637,0.251899438637,0,0,0,0,0,0,0,0,0,0,0,0,0,0.188346883469,0,0,0,0,0.12131242741,0,0.24262485482,0.162299167635,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0 |
| PF00005_ABC_tran | 137 | SM_ | 110 | 141 | 0.0377234744273,0.00461980790097,0.0096909780396,0.0157905225569,0.0169710021629,0.0275121349589,0.0156602458352,0.00405947701988,0.00739122457456,0.00768805945304,0,0.00161828107474,0.00591327371089,0.00202973850994,0.00579099946437,0.00416338695375,0.00323133939386,0,0.00548167659646,0.303901345366,0,0.280623915296,0.0067656772453,0.679542204351,0.697685738027,0.691214242456,0.00839368055968,0.00482361342933,0.0397878560196,0.0227702896963,0.00909516751323,0.0349315331438,0.0131520331553,0.0193392901203,0.0284420664734,0.0130227255943,0.012984391476,0.0106552933277,0.0204835408505,0.0189168490431,0.0285336657781,0.0275702117864,0,0.00457484438895,0,0,0,0.0238587624559,0.021829023946,0.00254510587901,0.019283918067,0,0.0217485765752,0.0237783150851,0.0213136565769,0.0276168909756,0.0118202358496,0.0114312928629,0.012052883022,0.0100482164063,0.0063542198101,0.00944680557652,0,0.00174289125479,0.0278798911495,0.097924307303,0.00184563007097,0.00635965898894,0.0322596343334,0.0297949758252,0.0490094392875,0.00790848545895,0.0292221420359,0.0249021779027,0,0.0234686984736,0.00517652112411,0.00215504189665,0,0,0.00764743755861,0.00471086684116,0.00909061002502,0.0225204772897,0.00473593873536,0,0.00399443207795,0.00197991580508,0,0,0.00270047617867,0.00251906964526,0.00289475535167,0.00277098703935,0.00270047617867,0.111023129603,0.0217327928995,0.019026506502,0.00270047617867,0.014177092878,0.0679676350364,0.0137430965532,0,0.190965797116,0,0.0585827481125,0.0127265974899,0.0652120185593,0.00794644519468,0.102944250852,0.0137201014491,0.0138465570152,0.0945003452098,0,0.0232318158738,0.00900928101363,0.00996736376179,0,0.0169296493927,0.018744119295,0.0136624119088,0.00214880200698,0.0166390770754,0.0058505131038,0.0142845969412,0.0157676346769,0.0166971239124,0.00307952606445,0,0,0.00264422779836,0.0129966476375,0.0352791479778,0.153867299413,0.00694183181232,0.00489649026725,0.0690208616258 |
| PF00005_ABC_tran | 137 | METABOLITE_ | 93 | 115 | 0.0423846282715,0.00244789601093,0.00981946362115,0.017596034204,0.0155837013976,0.0288244918514,0.0149514607241,0.00489579202186,0.00867883255515,0.00935790985627,0,0.00195760342646,0.00193456357756,0.00244789601093,0.00692361173264,0.00509590025931,0.00386912715512,0,0.00645575857075,0.248906074717,0,0.288270775052,0.00801153357221,0.663026833878,0.683136475548,0.681364675625,0.0102468162603,0.00641783224383,0.0346052146251,0.0257845741747,0.0109370965404,0.0417062809958,0.0158098487134,0.0232187100008,0.0311952501979,0.0155601075809,0.0136836831039,0.0109042741005,0.0224747866107,0.0219811610125,0.0265527236302,0.0273664044944,0,0.00558619284378,0,0,0,0.025763062536,0.023315166525,0.00313829683285,0.0201768696922,0,0.0201768696922,0.0226247657031,0.0226247657031,0.0290292426008,0.0110260155152,0.0135234901915,0.0144110449998,0.0119763190905,0.00752124098774,0.0111141050209,0,0.00207540533685,0.0302486042934,0.0524527668327,0.00227251781357,0.00765900406687,0.0354870010588,0.0354870010588,0.0526979917992,0.00665072854151,0.0292754942446,0.0269726888535,0,0.0252313733622,0.00629239146422,0.00260660765911,0,0,0.00886792919754,0.00555046745964,0.010788864225,0.0269824218852,0.00556363756128,0,0.00488547577917,0.00242060730148,0,0,0.00352968911807,0.00386606577791,0.00326618240823,0.00330305433987,0.00352968911807,0.115233025287,0.0130981412706,0.00653821951543,0.00352968911807,0.010863080338,0.0663599175597,0.0052259159312,0,0.184754657886,0,0.0599124302967,0.0162860401874,0.0573467317214,0.00555046745964,0.0728912112544,0.0135766150981,0.00528373197268,0.0739797676168,0,0.0166957068247,0.00427115563956,0.0118994288571,0,0.0185147541652,0.0118994288571,0.0147191112135,0.0026129579656,0.0179293823307,0.00330305433987,0.0154092075878,0.016929152311,0.0142435985256,0,0,0,0.00317755916369,0.0136260979711,0.0335947867361,0.121778722136,0.00840347509489,0.00584833317077,0.0677237376771 |
| PF00005_ABC_tran | 137 | ANP | 17 | 19 | 0,0,0.0235131301701,0,0,0.0235131301701,0.0470262603402,0,0,0,0,0,0,0,0,0,0,0,0,0.841006106374,0,0.271660563153,0,0.676289118674,0.676289118674,0.632069683936,0,0,0.0823856755492,0,0,0,0,0,0.031959449247,0,0.0235131301701,0.0235131301701,0.0235131301701,0.0470262603402,0.0940525206804,0.0705393905103,0,0,0,0,0,0,0,0,0,0,0,0,0,0.0470262603402,0,0,0,0,0,0,0,0,0,0.430099299531,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.102133756407,0.102133756407,0,0,0.062410917336,0.0597398340485,0,0.125591324241,0,0,0,0.117715952724,0.0255334391017,0.360214252687,0.0347094361003,0.0597398340485,0.303224405492,0,0.0597398340485,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.0442194347374,0.526216121352,0,0,0.229658360646 |
| PF00006_ATP-synt_ab | 213 | SM_ | 48 | 83 | 0,0,0,0.00418690801436,0,0,0.0203667663316,0,0.00386900178183,0,0,0.00386900178183,0,0,0,0.00386900178183,0,0,0,0,0,0,0.0988488771734,0.175445150161,0.0695603302947,0.00173400036405,0.57566732122,0.612799884263,0.638785090543,0.0886847595391,0.0538437941832,0.030641262013,0.0880153017934,0.0615367639825,0.0867187752413,0.0612003445172,0.0836536701719,0.0573313427354,0.0417983345638,0.0133194461894,0.0230254503209,0.019351851181,0.00386900178183,0.0339285033528,0.0242224992213,0.0121112496106,0,0,0,0,0.0739242998399,0.146139675162,0,0.00522074083795,0.0299399626262,0,0.0247466468246,0.0331204628534,0,0.00969600582193,0.00969600582193,0.0022941917051,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.00386900178183,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.0429700305796,0.0325605349484,0.0325605349484,0,0,0,0.0247466468246,0.0247466468246,0,0,0.0163728307959,0,0.00297268548145,0,0,0,0,0,0,0,0,0,0.00787881465062,0.00393940732531,0,0,0,0,0,0,0,0,0,0,0,0.00297268548145,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0.0459784485426,0,0,0.00173400036405,0,0,0,0,0.0125607240431,0.017819954343,0,0.0125607240431,0.0121859227816,0,0,0,0.0626004689813,0,0,0.00173400036405,0.0043609421676,0,0,0,0,0.0127797902401,0,0,0.0389426376458,0.199928757533,0.0125562590955,0.0022941917051,0.0117102485569,0.0177066053529,0.475712856261,0.00868954844585,0.014584989095,0.025337533588,0.0970702786178,0.0297709240626,0.045010602041,0.0189803777073,0.0471133143059,0.114051762747,0.0518791397563 |
| … | … | … | … | … | … |
You can see that the binding_frequencies column contains per residue scores from 0 to 1 that say
how likely each residue is to be involved in binding. Each value is comma separated and there are
as many values as there are match states in the HMM. That means the number of values in
binding_frequencies matches the value of domain_length. Also notice that the ligand_type varies
and you can have multiple ligands for a given Pfam. I hope its appreciated that these files give the essential output of
the InteracDome workflow, and there are two such files. The first is the non-redundant representable
set, which “correspond to domain-ligand interactions that had nonredundant instances across three
or more distinct PDB structures. [Kobren and Singh] recommend using this collection to learn more
about domain binding properties”. If I recall, there are 2375 such Pfam IDs in this set (this is
compared to the roughly 15,000-20,000 total Pfam IDs). The second set is the confident set, which
“correspond to domain-ligand interactions that had nonredundant instances across three or more
distinct PDB entries and achieved a cross-validated precision of at least 0.5. [Kobren and Singh]
recommend using this collection to annotate potential ligand-binding positions in protein
sequences”.
Description of the HMM profiles
Since each entry in the InteracDome dataset corresponds to a Pfam, we just need the Pfam database,
and that can be readily downloaded from the EBI ftp server, or by running anvi-setup-pfams. But
since all the Pfams are not present in the InteracDome dataset, we merely need a subset of the Pfam
database that contains Pfams with at least one entry from the non-redundant representable
InteracDome set. From now on I’ll just call this subset the InteracDome Pfams (IPfams). Also,
it needs to be said that InteracDome was carried out using Pfam 31.0, so it is critical that is
what we use too.
anvi-setup-interacdome
Acquiring the InteracDome’s tab-separated files and the IPfam HMM profiles are just one-time setups,
and therefore I created a program called anvi-setup-interacdome to handle these tasks. So to make these
inputs available, the user must run anvi-setup-interacdome which initiates a class called
InteracDomeSetup. Here is the class:
This is merely a snapshot of the class as of July 12th, 2020.
class InteracDomeSetup(object):
def __init__(self, args, run=terminal.Run(), progress=terminal.Progress()):
"""Setup a Pfam database for anvi'o
Parameters
==========
args : argparse.Namespace
See `bin/anvi-setup-interacdome` for available arguments
"""
self.run = run
self.progress = progress
self.interacdome_data_dir = args.interacdome_data_dir
self.pfam_setup = None
self.interacdome_files = {
'representable_interactions.txt': 'https://interacdome.princeton.edu/session/344807c477180230032a2a3807ba47c3/download/downloadBP?w=',
'confident_interactions.txt': 'https://interacdome.princeton.edu/session/344807c477180230032a2a3807ba47c3/download/downloadConfidentBP?w=',
}
if self.interacdome_data_dir and args.reset:
raise ConfigError("You are attempting to run InteracDome setup on a non-default data directory (%s) using the --reset flag. "
"To avoid automatically deleting a directory that may be important to you, anvi'o refuses to reset "
"directories that have been specified with --interacdome-data-dir. If you really want to get rid of this "
"directory and regenerate it with InteracDome data inside, then please remove the directory yourself using "
"a command like `rm -r %s`. We are sorry to make you go through this extra trouble, but it really is "
"the safest way to handle things." % (self.interacdome_data_dir, self.interacdome_data_dir))
if not self.interacdome_data_dir:
self.interacdome_data_dir = constants.default_interacdome_data_path
self.run.warning('', header='Setting up InteracDome', lc='yellow')
self.run.info('Data directory', self.interacdome_data_dir)
self.run.info('Reset contents', args.reset)
filesnpaths.is_output_dir_writable(os.path.dirname(os.path.abspath(self.interacdome_data_dir)))
if not args.reset and not anvio.DEBUG:
self.is_database_exists()
filesnpaths.gen_output_directory(self.interacdome_data_dir, delete_if_exists=args.reset)
def is_database_exists(self):
"""Raise ConfigError if database exists
Currently, this primitively decides if the database exists by looking at whether Pfam-A.hmm
or Pfam-A.hmm.gz exists.
"""
if (os.path.exists(os.path.join(self.interacdome_data_dir, 'Pfam-A.hmm') or
os.path.exists(os.path.join(self.interacdome_data_dir, 'Pfam-A.hmm.gz')))):
raise ConfigError("It seems you already have the InteracDome data downloaded in '%s', please "
"use --reset flag if you want to re-download it." % self.interacdome_data_dir)
def download_interacdome_files(self):
"""Download the confident and representable non-redundant InteracDome datasets
These datasets can be found at the interacdome webpage: https://interacdome.princeton.edu/
"""
for path, url in self.interacdome_files.items():
utils.download_file(
url,
os.path.join(self.interacdome_data_dir, path),
check_certificate=False,
progress=self.progress,
run=self.run
)
def download_pfam(self):
"""Setup the pfam data subset used by interacdome
Currently, interacdome only works for pfam version 31.0, so that is the version downloaded here.
"""
pfam_args = argparse.Namespace(
pfam_data_dir=self.interacdome_data_dir,
pfam_version='31.0',
reset=False,
)
self.pfam_setup = pfam.PfamSetup(pfam_args)
self.pfam_setup.get_remote_version()
self.pfam_setup.download(hmmpress_files=False)
def load_interacdome(self, kind='representable'):
"""Loads the representable interacdome dataset as pandas df"""
data = InteracDomeTableData(kind=kind, interacdome_data_dir=self.interacdome_data_dir)
return data.get_as_dataframe()
def get_interacdome_pfam_accessions(self):
"""Get the representable interacdome Pfam accessions"""
return set(self.load_interacdome(kind='representable').index.tolist())
def filter_pfam(self):
"""Filter Pfam data according to whether the ACC is in the InteracDome dataset"""
interacdome_pfam_accessions = self.get_interacdome_pfam_accessions()
hmm_profiles = pfam.HMMProfile(os.path.join(self.interacdome_data_dir, 'Pfam-A.hmm'))
hmm_profiles.filter(by='ACC', subset=interacdome_pfam_accessions)
hmm_profiles.write(filepath=None) # overwrites
# hmmpresses the new .hmm
self.pfam_setup.hmmpress_files()
# We also filter out the Pfam-A.clans.tsv file, since it is used as a catalog
clans_file = os.path.join(self.interacdome_data_dir, 'Pfam-A.clans.tsv')
clans = pd.read_csv(clans_file, sep='\t', header=None)
clans[clans[0].isin(interacdome_pfam_accessions)].to_csv(clans_file, sep='\t', index=False, header=False)
def setup(self):
"""The main method of this class. Sets up the InteracDome data directory for usage"""
self.run.warning('', header='Downloading InteracDome tables', lc='yellow')
self.download_interacdome_files()
self.run.warning('', header='Downloading associated Pfam HMM profiles', lc='yellow')
self.download_pfam()
self.run.warning('', header='Filtering Pfam HMM profiles', lc='yellow')
self.filter_pfam()
First, anvi’o creates a directory under anvio/data/misc/InteracDome (by default) which will end up
housing the IPfam HMM profiles and the InteracDome datasets. Then the main method, setup is
called, which is directly above us. We can see that first, anvi’o downloads the tab-separated files.
Since these files were created for Pfam version 31.0, anvi’o next downloads a copy of the Pfam
31.0 HMMs. There already exists a framework to download Pfams because of anvi-setup-pfams, so
this was very little work to do. Yay for code resusability. As a final step, the Pfam HMMs are
subset to the IPfam HMMs, i.e. only the Pfams that are in the InteracDome dataset.
(3) Running the hidden Markov model (HMM)
HMMs are by no means an elementary topic, and so rather than butcher an explanation with my limited understanding, I defer to this wonderful paper.
With both the required inputs being gathered and setup with anvi-setup-interacdome, the next thing I focused on was actually
running an HMM on user genes. This as well as the remainder of steps will be carried out by the
program anvi-run-interacdome.
The program that goes hand-in-hand with Pfam HMM profiles is
HMMER, and in fact, anvi’o already has a driver
to run HMMER on a set of user genes which was written for anvi-run-pfams. Further in fact(?), anvi-run-pfams in some ways does exactly what I want: it takes
genes from the contigs database, runs an HMM of the user genes against the Pfam HMM profiles,
and annotates genes with the best Pfam hit. The primary difference between
anvi-run-interacdome and
anvi-run-pfams, is that rather than
simply attribute to each gene the Pfam ID of the best hit, we instead must keep track of
residue-level information associated to each hit, so that we can associate the binding
frequencies from the InteracDome dataset. In this sense, things are much more complicated, as I will
need to write an in-depth parser of HMMER’s output to keep track of these things.
Currently, anvi-run-pfams can be
run with either of the HMMER programs, hmmscan or hmmsearch. However, for good
reason,
hmmsearch in practice runs much faster for querying user genes against a HMM profile database with
a size comparable to the Pfam database. Since hmmsearch is faster for our use-case and because I
wanted to avoid creating 2 output parsers (one for hmmscan and one for hmmsearch),
anvi-run-interacdome is by design only compatible with hmmsearch.
All in all, anvi-run-interacdome runs hmmsearch almost exactly like
anvi-run-pfams does. In fact,
the responsible class for anvi-run-interacdome, anvio.interacdome.InteracDomeSuper,
inherits anvio.pfam.Pfam to manage a lot of the boiler-plate code.
I chose to apply the HMMER filter parameter --cut_ga that uses the Pfam GA (gathering threshold)
score cutoffs to pre-filter the output of hmmsearch. This cuts out a lot of crap to sift through.
(4) Parsing HMMER’s standard output file
hmmsearch has 2 outputs. One is a tabular output like this:
# --- full sequence --- -------------- this domain ------------- hmm coord ali coord env coord
# target name accession tlen query name accession qlen E-value score bias # of c-Evalue i-Evalue score bias from to from to from to acc description of target
#------------------- ---------- ----- -------------------- ---------- ----- --------- ------ ----- --- --- --------- --------- ------ ----- ----- ----- ----- ----- ----- ----- ---- ---------------------
3609 - 333 2-Hacid_dh PF00389.29 134 8.8e-20 68.1 0.1 1 1 8.7e-23 1.2e-19 67.7 0.1 9 133 20 325 8 326 0.95 -
5374 - 320 2-Hacid_dh PF00389.29 134 2e-10 37.8 0.0 1 1 2.6e-13 3.6e-10 37.0 0.0 21 103 24 195 5 310 0.71 -
3609 - 333 2-Hacid_dh_C PF02826.18 178 1e-55 185.2 0.0 1 1 1.6e-58 1.5e-55 184.7 0.0 2 177 118 293 117 294 0.98 -
5374 - 320 2-Hacid_dh_C PF02826.18 178 1.7e-53 178.0 0.1 1 1 3.2e-56 2.9e-53 177.2 0.1 2 178 108 282 107 282 0.96 -
3608 - 311 2-Hacid_dh_C PF02826.18 178 2.4e-07 27.7 0.0 1 1 4.4e-10 4e-07 26.9 0.0 36 144 12 123 5 130 0.85 -
2301 - 162 2Fe-2S_thioredx PF01257.18 145 3.8e-37 124.7 0.2 1 1 1.6e-40 4.3e-37 124.5 0.2 4 144 12 154 9 155 0.95 -
2121 - 441 2HCT PF03390.14 416 8.5e-157 519.4 35.0 1 1 3.6e-160 9.9e-157 519.2 35.0 1 415 24 434 24 435 0.99 -
1998 - 329 3Beta_HSD PF01073.18 282 3.1e-22 76.2 0.0 1 1 1.1e-23 3.9e-21 72.6 0.0 1 237 4 243 4 254 0.74 -
504 - 316 3Beta_HSD PF01073.18 282 7.7e-18 61.8 0.1 1 1 3.4e-20 1.2e-17 61.2 0.1 1 229 5 232 5 242 0.78 -
[...]
The format is tabularized and thus easy enough to parse, but unfortunately the information is not detailed enough since it contains no information about residue-by-residue alignment of the match states (residues in the HMM profile) to the user gene sequence. The second, more verbose output, does:
# hmmsearch :: search profile(s) against a sequence database
# HMMER 3.2.1 (June 2018); http://hmmer.org/
# Copyright (C) 2018 Howard Hughes Medical Institute.
# Freely distributed under the BSD open source license.
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
# query HMM file: /Users/evan/Software/anvio/anvio/data/misc/InteracDome/Pfam-A.hmm
# target sequence database: /var/folders/58/mpjnklbs5ql_y2rsgn0cwwnh0000gn/T/tmpyebp3gbo/AA_gene_sequences.fa.0
# output directed to file: /var/folders/58/mpjnklbs5ql_y2rsgn0cwwnh0000gn/T/tmpyebp3gbo/AA_gene_sequences.fa.0_output
# per-dom hits tabular output: /var/folders/58/mpjnklbs5ql_y2rsgn0cwwnh0000gn/T/tmpyebp3gbo/AA_gene_sequences.fa.0_table
# model-specific thresholding: GA cutoffs
# number of worker threads: 1
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Query: 14-3-3 [M=222]
Accession: PF00244.19
Description: 14-3-3 protein
Scores for complete sequences (score includes all domains):
--- full sequence --- --- best 1 domain --- -#dom-
E-value score bias E-value score bias exp N Sequence Description
------- ------ ----- ------- ------ ----- ---- -- -------- -----------
[No hits detected that satisfy reporting thresholds]
Domain annotation for each sequence (and alignments):
[No targets detected that satisfy reporting thresholds]
Internal pipeline statistics summary:
-------------------------------------
Query model(s): 1 (222 nodes)
Target sequences: 2754 (847468 residues searched)
Passed MSV filter: 207 (0.0751634); expected 55.1 (0.02)
Passed bias filter: 80 (0.0290487); expected 55.1 (0.02)
Passed Vit filter: 6 (0.00217865); expected 2.8 (0.001)
Passed Fwd filter: 0 (0); expected 0.0 (1e-05)
Initial search space (Z): 2754 [actual number of targets]
Domain search space (domZ): 0 [number of targets reported over threshold]
# CPU time: 0.01u 0.00s 00:00:00.01 Elapsed: 00:00:00.01
# Mc/sec: 14884.77
//
Query: 2-Hacid_dh [M=134]
Accession: PF00389.29
Description: D-isomer specific 2-hydroxyacid dehydrogenase, catalytic domain
Scores for complete sequences (score includes all domains):
--- full sequence --- --- best 1 domain --- -#dom-
E-value score bias E-value score bias exp N Sequence Description
------- ------ ----- ------- ------ ----- ---- -- -------- -----------
8.8e-20 68.1 0.1 1.2e-19 67.7 0.1 1.1 1 3609
2e-10 37.8 0.0 3.6e-10 37.0 0.0 1.5 1 5374
Domain annotation for each sequence (and alignments):
>> 3609
# score bias c-Evalue i-Evalue hmmfrom hmm to alifrom ali to envfrom env to acc
--- ------ ----- --------- --------- ------- ------- ------- ------- ------- ------- ----
1 ! 67.7 0.1 8.7e-23 1.2e-19 9 133 .. 20 325 .. 8 326 .. 0.95
Alignments for each domain:
== domain 1 score: 67.7 bits; conditional E-value: 8.7e-23
HHHHHHHHH.TTE.EEEEESCSSHHHCHHHHTTESEEEE-TTS-BSHHHHHHHTT--EEEESSSSCTTB-HHHHHHTT-EEEE-TT.TTHHHHHHHH.. CS
xxxxxxxxx.xxx.xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx.xxxxxxxxxx.. RF
2-Hacid_dh 9 eeelellke.egv.evevkdellteellekakdadalivrsntkvtaevlealpkLkviaragvGvDnvDldaakerGilVtnvpg.ystesvAElt.. 102
+e l +l++ ++v +++v +e+ +el+e ++++ ++i ++ +t+e++e+ ++L +i+r+g+G++n+Dldaak++ +V+ +p+ + ++vAE
3609 20 PEHLTRLEKiGTVkHFTVDSEIGGKELAECLQGYTIIIASVTPFFTKEFFEHKDELLLISRHGIGYNNIDLDAAKQHDTIVSIIPAlVERDAVAENNvt 118
667777888655556777779*****************************************************************777889***999* PP
................................................................................................... CS
................................................................................................... RF
2-Hacid_dh 103 ................................................................................................... 102
3609 119 nllavlrqtvaadasvkadqwekranfvgrtlfnktvgvigvgntgscvvetlrngfrcdvlaydpyksatylqsygakkvdldtllasadiiclcanl 217
*************************************************************************************************** PP
.............................................................................T-BHHHHHHHHHHHHHHHHHHH CS
.............................................................................xxxxxxxxxxxxxxxxxxxxxx RF
2-Hacid_dh 103 .............................................................................aaTeeaqeniaeeaaenlvafl 124
a+T e +++++e++++ +++++
3609 218 teesyhmigsaeiakmkdgvylsnsargalideeamiaglqsgkiaglgtdvleeepgrknhpylafenvvmtphtsAYTMECLQAMGEKCVQDVEDVV 316
*************************************************************************************************** PP
TTCCGTTBC CS
xxxxxxxxx RF
2-Hacid_dh 125 kgespanav 133
+g p+ v
3609 317 QGILPQRTV 325
**7777655 PP
>> 5374
# score bias c-Evalue i-Evalue hmmfrom hmm to alifrom ali to envfrom env to acc
--- ------ ----- --------- --------- ------- ------- ------- ------- ------- ------- ----
1 ! 37.0 0.0 2.6e-13 3.6e-10 21 103 .. 24 195 .. 5 310 .. 0.71
Alignments for each domain:
== domain 1 score: 37.0 bits; conditional E-value: 2.6e-13
EEEEESCSSHHHCHHHHTT.ESEEEE-TTS-BSHHHHHHH.TT--EEEESSSSCTTB-HHHHHHTT-EEEE-TTTTHHHHHHHH............... CS
xxxxxxxxxxxxxxxxxxx.xxxxxxxxxxxxxxxxxxxx.xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx............... RF
2-Hacid_dh 21 evevkdellteellekakd.adalivrsntkvtaevleal.pkLkviaragvGvDnvDldaakerGilVtnvpgystesvAElt............... 102
++ + +t+ l ++ ++ +++++ + +++ ++l+ + Lk+i+++++G D D+d ++e+Gil++n g ++ s+ E++
5374 24 APNYLVKTSTDYLSSAEEEaIEIMLGWH-KEIGPRLLASDtSHLKWIQLISAGADYMDFDKLREKGILLSNGSGIHSVSISEHVlgvllahtrglqesi 121
4444446666655555555344555555.559999998888************************************************9999888888 PP
.........................................................................T CS
.........................................................................x RF
2-Hacid_dh 103 .........................................................................a 103
5374 122 qqqmqhtwnqtapsyqqlsgqkmlivgtgqigqqlakfakglnlqvygvntsghvtegfiecysqknmskiihE 195
88888888888888888888888888888888888888888888888887777777775554444444444440 PP
[...]
To write the parser I made extensive use of the detailed output description in the HMMER user guide (search for “Step 2: search the sequence database with hmmsearch”) for the relevant section. I won’t reiterate the meaning of each line, so check the guide if you’re keen. The most important thing to note is that this more verbose output contains the same tabular information as the first output (albeit in more scattered fashion), and additionally has alignments of the HMM profile to the query gene.
Consider this section of the output:
Query: 2-Hacid_dh [M=134]
Accession: PF00389.29
Description: D-isomer specific 2-hydroxyacid dehydrogenase, catalytic domain
Scores for complete sequences (score includes all domains):
--- full sequence --- --- best 1 domain --- -#dom-
E-value score bias E-value score bias exp N Sequence Description
------- ------ ----- ------- ------ ----- ---- -- -------- -----------
8.8e-20 68.1 0.1 1.2e-19 67.7 0.1 1.1 1 3609
2e-10 37.8 0.0 3.6e-10 37.0 0.0 1.5 1 5374
I will refer to this as the domain hits summary. In this case, the IPfam PF00389.29 hit to 2 user genes: 3609 and 5374. Then, for each of these
genes, we can find further down in the file the alignment of each. For example, here is where the
alignment info for gene 3609 starts:
>> 3609
# score bias c-Evalue i-Evalue hmmfrom hmm to alifrom ali to envfrom env to acc
--- ------ ----- --------- --------- ------- ------- ------- ------- ------- ------- ----
1 ! 67.7 0.1 8.7e-23 1.2e-19 9 133 .. 20 325 .. 8 326 .. 0.95
Alignments for each domain:
== domain 1 score: 67.7 bits; conditional E-value: 8.7e-23
HHHHHHHHH.TTE.EEEEESCSSHHHCHHHHTTESEEEE-TTS-BSHHHHHHHTT--EEEESSSSCTTB-HHHHHHTT-EEEE-TT.TTHHHHHHHH.. CS
xxxxxxxxx.xxx.xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx.xxxxxxxxxx.. RF
2-Hacid_dh 9 eeelellke.egv.evevkdellteellekakdadalivrsntkvtaevlealpkLkviaragvGvDnvDldaakerGilVtnvpg.ystesvAElt.. 102
+e l +l++ ++v +++v +e+ +el+e ++++ ++i ++ +t+e++e+ ++L +i+r+g+G++n+Dldaak++ +V+ +p+ + ++vAE
3609 20 PEHLTRLEKiGTVkHFTVDSEIGGKELAECLQGYTIIIASVTPFFTKEFFEHKDELLLISRHGIGYNNIDLDAAKQHDTIVSIIPAlVERDAVAENNvt 118
667777888655556777779*****************************************************************777889***999* PP
[...]
I will refer to this as the hit alignment. The aggregation of all the domain hit summaries and the
hit alignments are stored in two datastructures that are attributes of the anvio.parsers.hmmer.HMMERStandardOutput
parser class. I will now describe both of them:
The first is dom_hits, which is a dataframe which looks like this:
| pfam_name | pfam_id | corresponding_gene_call | domain | qual | score | bias | c-evalue | i-evalue | hmm_start | hmm_stop | hmm_bounds | ali_start | ali_stop | ali_bounds | env_start | env_stop | env_bounds | mean_post_prob | match_state_align | comparison_align | sequence_align | version | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Beta_elim_lyase | PF01212 | 1762 | 1 | ! | 20.9 | 0.1 | 1e-08 | 3.5e-06 | 33 | 169 | .. | 44 | 177 | .. | 34 | 215 | .. | 0.72 | tvnrLedavaelfgke..aalfvpqGtaAnsill.kill.qr..geevivtepahihfdetgaiaelagvklrdlknkeaGkmdlekleaaikevgaheekiklisltvTnntagGqvvsleelrevaaiakkygiplhlDgA | ++ +++ael+ + f+ Gt +++ l + + +r g+ +i++ h +et + g +l ++ +++G +++e+l+++i++ e i + +++v n+ G++ +++e+ ev +a+ +i++h+D+ | LLQQARKQIAELINVSanEIYFTSGGTEGDNWVLkGTAIeKRefGNHIIISAVEHPAVTETAEQLVELGFELSYAPVDKEGRVKVEELQKLIRK—–ETILVSVMAVNNE–VGTIQPIKEISEV–LAEFPKIHFHVDAV | 20 |
| 1 | PAPS_reduct | PF01507 | 1541 | 1 | ! | 36.1 | 0.1 | 3.6e-13 | 1.3e-10 | 2 | 164 | .. | 21 | 231 | .. | 20 | 234 | .. | 0.79 | lvvsvsgGkdslVllhLalkafkpv….pvvfvdtghefpetiefvdeleeryglrlkvyepeeevaekinaekhgs.slyee.aaeriaKveplkk……………………………aLekldedall..tGaRrdesksraklpiveidedfek………slrvfPllnWteedvwqyilrenipynpLydqgfr | + +s+sgGkds +++La + ++ ++ ++ + ++ t++f++++e+ +++ +++ ++++ + + +++ + + + e+ + p k e++ ++a+ +G+R++es +r++ +++ +++ + ++Pl++W+ d+w+ + +++yn +y++ ++ | VYFSFSGGKDSGLMVQLANLVAEKLdrnfDLLILNIEANYTATVDFIKKIEQLPRVKNIYHFCLPFFEDNNTSFFQPQwKMWDPsEKEKWIHSLP–KnaitleniddglkkyyslsngnpdrflryfqnwYKEQYPQSAIScgVGIRAQESLHRHSAVTKGENKYKNRcwinitlegNILFYPLFDWKVGDIWAATFKCELEYNYIYEKMYK | 18 |
| 2 | Ank_2 | PF12796 | 1756 | 1 | ! | 32.2 | 0 | 6.7e-12 | 2.3e-09 | 29 | 84 | .] | 74 | 135 | .. | 53 | 135 | .. | 0.85 | aLhyAakngnleivklLle…h.a..adndgrtpLhyAarsghleivklLlekgadinlkd | aL Aa + +++ vk +l+ + + +d +g+tpL +A+ ++ +ei+k L+++gadinl++ | ALLEAANQRDTKKVKEILQdttYqVdeVDTEGNTPLNIAVHNNDIEIAKALIDRGADINLQN | 6 |
| 3 | Ank_2 | PF12796 | 1756 | 2 | ! | 28.5 | 0 | 9.5e-11 | 3.3e-08 | 22 | 75 | .. | 199 | 265 | .. | 195 | 267 | .] | 0.76 | pn..k.ngktaLhyAak..ngnl…eivklLleha…..adndgrtpLhyAarsghleivklLle | ++ + +g taL+ A+ +gn +ivklL+e++ dn+grt++ yA ++g++ei k+L + | IDfqNdFGYTALIEAVGlrEGNQlyqDIVKLLMENGadqsiKDNSGRTAMDYANQKGYTEISKILAQ | 6 |
| 4 | IGPS | PF00218 | 1615 | 1 | ! | 20.6 | 0.1 | 1.2e-08 | 4e-06 | 202 | 249 | .. | 195 | 242 | .. | 73 | 248 | .. | 0.88 | LaklvpkdvllvaeSGiktredveklkeegvnafLvGeslmrqedvek | +++lv+++++++ae i+t+e+++++k+ gv ++ vG +++r ++ +k | IKQLVQENICVIAEGKIHTPEQARQIKKLGVAGIVVGGAITRPQEIAK | 20 |
| 5 | Ribosomal_L33 | PF00471 | 1562 | 1 | ! | 66.6 | 1.5 | 1.1e-22 | 3.7e-20 | 2 | 47 | .] | 4 | 49 | .] | 3 | 49 | .] | 0.97 | kvtLeCteCksrnYtttknkrntperLelkKYcprcrkhtlhkEtK | +++LeC e+++r Y t+knkrn+perLelkKY p++r++ ++kE K | NIILECVETGERLYLTSKNKRNNPERLELKKYSPKLRRRAIFKEVK | 19 |
| 6 | Ribosomal_S14 | PF00253 | 1565 | 1 | ! | 83.3 | 0.1 | 3.9e-28 | 1.3e-25 | 2 | 54 | .] | 36 | 88 | .. | 35 | 88 | .. | 0.98 | laklprnssptrirnrCrvtGrprGvirkfgLsRicfRelAlkgelpGvkKaS | laklpr+s+p+r+r r++ +GrprG++rkfg+sRi+fRel ++g +pGvkKaS | LAKLPRDSNPNRLRLRDQTDGRPRGYMRKFGMSRIKFRELDHQGLIPGVKKAS | 20 |
| 7 | Polysacc_synt_C | PF14667 | 1593 | 1 | ! | 61.4 | 19.2 | 5.4e-21 | 1.9e-18 | 2 | 139 | .. | 371 | 516 | .. | 370 | 519 | .. | 0.83 | LailalsiiflslstvlssiLqglgrqkialkalvigalvklilnllliplfgivGaaiatvlallvvavlnlyalrrllgikl…llrrllkpllaalvmgivvylllllllglllla…al..alllavlvgalvYllllll | L+ ++s+ +l+++t++ siLq+l +k+a+ ++ i++l+kli+++++i+lf +G +iat+++ ++++++ +++l+r++ i++ ++ +++ +++vm i+ +l+l+++ ++ + +l + l +++g++v+ + l++ | LSATIISTSLLGIFTIVLSILQALSFHKKAMQITSITLLLKLIIQIPCIYLFKGYGLSIATIICTMFTTIIAYRFLSRKFDINPikyNRKYYSRLVYSTIVMTILSLLMLKIISSVYKFEstlQLffLISLIGCLGGVVFSVTLFR | 5 |
It stores all the domain hit information in the domain hit summaries as well as the sequence of the
consensus match states, the comparison string, and the sequence of the user gene. This table
contains all essential information, however the alignment information is in the form of raw strings
(see match_state_align, comparison_align, and sequence_align columns).
To further process the alignment info into a more meaningful form, there is a second data structure
called ali_info. ali_info is a nested dictionary. It looks like this:
ali_info = {
# here is the template
<gene_callers_id>: {
(<pfam_id>, <domain_id>): {
<a_dataframe_with_alignment_info>
},
},
# here is an example
1699: {
('PF07972', 1): {
<a_dataframe_with_alignment_info>
},
('PF07972', 2): {
<another_dataframe_with_alignment_info>
},
('PF03383', 1): {
<yet_another_dataframe_with_alignment_info>
},
},
}
Each dataframe contains detailed alignment info for a given hit. In the above example, if we wanted
to access alignment info about the hit of PF00389.29 against the user gene with ID 3609, we
would access the dataframe via ali_info[3609][('PF00389', 1)]. Since PF00389 hit only once to
3609, the hit has the domain ID 1. This entry in the nested dictionary is a dataframe that looks
like this:
| seq | hmm | comparison | seq_positions | hmm_positions | |
|---|---|---|---|---|---|
| 0 | P | E | + | 19 | 8 |
| 1 | E | E | E | 20 | 9 |
| 2 | H | E | 21 | 10 | |
| 3 | L | L | L | 22 | 11 |
| 4 | T | E | 23 | 12 | |
| 5 | R | L | + | 24 | 13 |
| 6 | L | L | L | 25 | 14 |
| 7 | E | K | + | 26 | 15 |
This dataframe is a per-residue characterization of the alignment strings, i.e. the columns
match_state_align, sequence_align, and comparison_align found in dom_hits. For convenience,
positions in the alignment that contain a gap either in the HMM or in the gene sequence are stripped
from this dataset. seq_positions correspond to the 0-indexed positions in the user gene (in this
example the gene is 3609). In other words, these are the codon_order_in_gene values that aligned to a non-gap
position in the HMM. Why doesn’t seq_positions start at 0? Because in this example, the hit starts
at the 20th amino acid in the user gene. Sanity check: this means the ali_start entry in the
corresponding row of self.dom_hits would be 20.
Together, these 2 data structures are the fruits of labor from parsing the HMMER standard out and
provide generic utility that extends beyond just anvi-run-interacdome.
(5) Filtering HMM hits
Pfam gathering threshold
As previously mentioned, hmmsearch is ran with the --cut_ga flag, which uses Pfam gathering
thresholds to pre-filter a lot of poor-quality hits.
Filtering partial hits
In the InteracDome paper, Kobren and Singh are more stringent about filtering hits than I decided to be. In the paper, it is demanded that 100% of the Pfam HMM maps to the user gene, i.e. the first and last position of the HMM hit should be non-gap characters, or else the hit is discarded. In other words, they discarded all partial hits. I have opted to relax this constraint since it seemed too strict for my applications, however, I did want to enforce some kind of threshold that would discard hits that are very partial. To quantify the partialness of a hit I defined the hit fraction quantity, which is defined as
\[f = K/L\]where $K$ is the alignment length (hmm_stop - hmm_start) and $L$ is the length of the HMM
profile (the number of match states). For example, in the hit below, PF01212 has length of $293$
(not shown below, but trust me), and the alignment length is $169-33=136$. This means the hit fraction is
$f = 136/293 \approx 0.46$.
| pfam_name | pfam_id | corresponding_gene_call | domain | qual | score | bias | c-evalue | i-evalue | hmm_start | hmm_stop | hmm_bounds | ali_start | ali_stop | ali_bounds | env_start | env_stop | env_bounds | mean_post_prob | match_state_align | comparison_align | sequence_align | version | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Beta_elim_lyase | PF01212 | 1762 | 1 | ! | 20.9 | 0.1 | 1e-08 | 3.5e-06 | 33 | 169 | .. | 44 | 177 | .. | 34 | 215 | .. | 0.72 | tvnrLedavaelfgke..aalfvpqGtaAnsill.kill.qr..geevivtepahihfdetgaiaelagvklrdlknkeaGkmdlekleaaikevgaheekiklisltvTnntagGqvvsleelrevaaiakkygiplhlDgA | ++ +++ael+ + f+ Gt +++ l + + +r g+ +i++ h +et + g +l ++ +++G +++e+l+++i++ e i + +++v n+ G++ +++e+ ev +a+ +i++h+D+ | LLQQARKQIAELINVSanEIYFTSGGTEGDNWVLkGTAIeKRefGNHIIISAVEHPAVTETAEQLVELGFELSYAPVDKEGRVKVEELQKLIRK—–ETILVSVMAVNNE–VGTIQPIKEISEV–LAEFPKIHFHVDAV | 20 |
To apply a filter based on partialness, I created a threshold value, self.min_hit_threshold
(specified with --min-hit-fraction) that the hit fraction of a hit must exceed in order to be
kept. If self.min_hit_threshold = 0.5, then the above hit would be removed since its hit fraction
is less than this value ($\approx 0.46$). To determine a reasonable default value for this threshold, I calculated the
hit fractions for all hits of an E. faecalis genome against the IPfams which yielded 2898 hits.
Here is the resulting histogram of hit fractions:
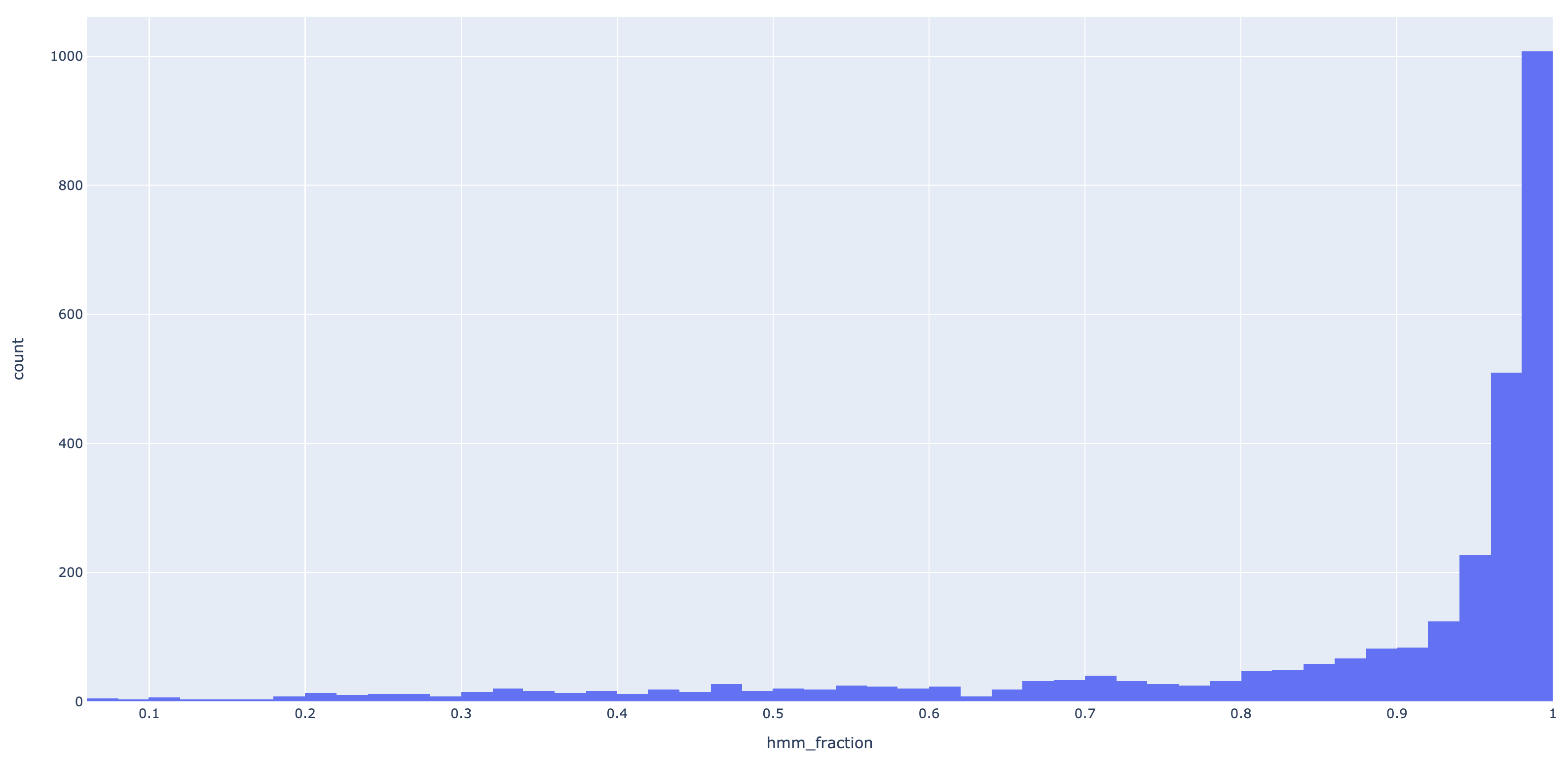
From this histogram, I deemed 0.8 was a good compromise between data retention and quality, so that is the default on July 21st, 2020.
Filtering bad hits with information content
Really, filtering bad hits is what the Pfam gathering threshold is designed for. But to be extra sure that we are not including junk, I followed the protocol of the InteracDome paper. To understand what Kobren and Singh did, we must first make a one paragraph detour to define some terms.
An HMM profile is composed of match states, one for each residue position in the HMM profile. And each match state is composed of 20 emission probabilities, one for each amino acid. An emission probability gives a probability that a given amino acid is observed for the match state. So match states which are highly conserved will have a single dominant emission probability, whereas an unconserved match state will have more uniformly distributed emission probabilities, meaning many amino acids can inhabit the match state. To quantify how conserved a match state is, there is a quantity used in sequence logos called information content (IC), which compares the entropy of a perfectly uniform distribution of emission probabilities to the observed entropy of emission probabilities. The formula for IC boils down to:
\[\text{IC} = \log_2{20} + \sum_{i=1}^{20}{ f_i \log_2{f_i} }\]where $f_i$ is the emission probability of the $i$th amino acid. The equation’s worth boils down to this: the higher the information content, the more conserved the match state is.
With this in mind, Kobren and Singh identified each match state in a hit that had an IC exceeding 4 (very conserved) and took note of the consensus amino acid (the amino acid with the highest emission probability). Then, in order to retain a hit, the gene sequence this HMM profile hit to must share all the same amino acids as the consensus amino acids at each of these conserved positions. The idea is that if one is to trust the quality of the hit, these positions should equate since they are conserved. I wanted to replicate this filtering procedure.
This is great and all, except the HMMER output does not provide emission probabilities, and therefore calculating IC is a non-trivial task…
Calculating information content
To calculate information content (IC), one must dive into the .hmm file itself (the one created
during anvi-setup-interacdome). An .hmm file contains all the information necessary about each
HMM profile. Here is the content for just one HMM profile in the .hmm:
HMMER3/f [3.1b2 | February 2015]
NAME SnoaL_2
ACC PF12680.6
DESC SnoaL-like domain
LENG 102
ALPH amino
RF no
MM no
CONS yes
CS yes
MAP yes
DATE Fri Jan 20 17:09:36 2017
NSEQ 346
EFFN 15.065170
CKSUM 2037425494
GA 27.00 27.00;
TC 27.00 27.00;
NC 26.90 26.90;
BM hmmbuild HMM.ann SEED.ann
SM hmmsearch -Z 26740544 -E 1000 --cpu 4 HMM pfamseq
STATS LOCAL MSV -9.2504 0.71708
STATS LOCAL VITERBI -10.2672 0.71708
STATS LOCAL FORWARD -3.5453 0.71708
HMM A C D E F G H I K L M N P Q R S T V W Y
m->m m->i m->d i->m i->i d->m d->d
COMPO 2.34288 4.65458 2.72525 2.62519 3.03686 2.67067 3.52524 3.01724 3.00945 2.64165 3.76982 3.29469 3.35980 3.23149 2.70953 2.90053 2.80605 2.55611 4.08669 3.52142
2.68618 4.42225 2.77519 2.73123 3.46354 2.40513 3.72494 3.29354 2.67741 2.69355 4.24690 2.90347 2.73739 3.18146 2.89801 2.37887 2.77519 2.98518 4.58477 3.61503
0.05491 6.34906 2.96263 0.61958 0.77255 0.00000 *
1 1.85458 4.97177 5.25772 5.11150 3.08317 4.68531 4.99660 2.29317 4.89333 2.21381 3.63812 4.70077 5.28514 5.02200 4.89677 3.94172 3.72771 0.88269 4.66437 3.70054 1 v - - H
2.68618 4.42225 2.77519 2.73123 3.46354 2.40513 3.72494 3.29354 2.67741 2.69355 4.24690 2.90347 2.73739 3.18146 2.89801 2.37887 2.77519 2.98518 4.58477 3.61503
0.00274 6.29689 7.01924 0.61958 0.77255 0.61121 0.78240
2 2.68388 5.69592 2.79379 1.70132 5.25800 3.51515 3.94249 4.36550 2.81371 3.15390 5.18636 3.57935 4.80095 2.41963 1.52011 3.04363 2.67472 3.24049 6.57819 4.76611 2 r - - H
2.68618 4.42225 2.77519 2.73123 3.46354 2.40513 3.72494 3.29354 2.67741 2.69355 4.24690 2.90347 2.73739 3.18146 2.89801 2.37887 2.77519 2.98518 4.58477 3.61503
0.00268 6.31868 7.04103 0.61958 0.77255 0.65639 0.73131
3 1.69557 6.15246 2.61454 2.25344 4.80524 3.13834 3.50575 4.98574 2.35803 3.99893 4.81684 3.13184 4.80553 2.73142 1.63172 3.26000 2.93426 4.54380 6.58436 4.54785 3 r - - H
2.68618 4.42225 2.77519 2.73123 3.46354 2.40513 3.72494 3.29354 2.67741 2.69355 4.24690 2.90347 2.73739 3.18146 2.89801 2.37887 2.77519 2.98518 4.58477 3.61503
0.00981 6.32551 4.83198 0.61958 0.77255 0.48312 0.95934
[... removed this part to keep things reasonably sized ...]
100 1.71561 3.64865 4.38480 3.42760 4.07808 4.69231 4.80409 3.24425 2.74698 3.28918 4.57912 4.05114 5.07262 3.40708 2.94600 2.98074 1.91959 1.46391 4.53056 4.17751 268 v - - E
2.68618 4.42225 2.77519 2.73123 3.46354 2.40513 3.72494 3.29354 2.67741 2.69355 4.24690 2.90347 2.73739 3.18146 2.89801 2.37887 2.77519 2.98518 4.58477 3.61503
0.01668 6.34179 4.21465 0.61958 0.77255 0.56154 0.84473
101 2.40957 4.62174 3.16892 1.34079 4.55620 3.69164 2.87511 4.51265 2.91940 2.76532 5.18494 3.33723 4.80964 3.03069 2.08808 2.60615 3.78804 3.55867 4.81816 3.79971 269 e - - E
2.68618 4.42225 2.77519 2.73123 3.46354 2.40513 3.72494 3.29354 2.67741 2.69355 4.24690 2.90347 2.73739 3.18146 2.89801 2.37887 2.77519 2.98518 4.58477 3.61503
0.05884 6.32777 2.89402 0.61958 0.77255 0.69190 0.69440
102 2.66258 5.17554 4.55953 2.87507 3.15074 3.58098 2.17249 2.34555 3.60158 2.58045 3.32878 3.02642 5.07290 3.26130 3.35997 3.29569 3.03481 2.15305 2.79882 3.02577 270 v - - E
2.68618 4.42225 2.77519 2.73123 3.46354 2.40513 3.72494 3.29354 2.67741 2.69355 4.24690 2.90347 2.73739 3.18146 2.89801 2.37887 2.77519 2.98518 4.58477 3.61503
0.00189 6.27083 * 0.61958 0.77255 0.00000 *
//
The important part for IC is the section with lines that start from 1 and go to 102. Each of
these numbers are the match states of the HMM profile–in this case the profile has 102 match
states. On each line starting with one of these numbers are the negative natural log of the emission
probabilities for each of the 20 amino acids. Therefore, by parsing this file by taking the negative
exponentiation of these values, one can establish the
emission probabilities for every match state in every HMM profile.
To grab the IC values for each match state, I wrote a parser class, anvio.pfam.HMMProfile, that
parses .hmm profiles. I tried to make something generic, not just for the single-minded purpose of
calculating IC, so it is basic but also extensible for future purposes. From what I can tell, most
of the information in basic .hmm formats is being captured. Alongside this, IC is calculated for
each match state in a dataframe that can be accessed like so:
print(anvio.pfam.HMMProfile(<filepath>).data['PF01965']['MATCH_STATES'])
This prints the following:
| MATCH_STATE | CS | MM | RF | CONS | MAP | IC |
|---|---|---|---|---|---|---|
| 0 | E | - | - | K | 1 | 0.469514 |
| 1 | E | - | - | K | 2 | 0.992492 |
| 2 | E | - | - | V | 3 | 1.79484 |
| 3 | E | - | - | L | 4 | 0.83366 |
| 4 | E | - | - | V | 5 | 1.30396 |
| 5 | E | - | - | L | 6 | 0.758542 |
| 6 | E | - | - | L | 7 | 0.779465 |
| … | … | … | … | … | … | … |
| 148 | E | - | - | S | 214 | 1.88744 |
| 149 | S | - | - | R | 215 | 0.457319 |
| 150 | S | - | - | G | 216 | 0.439365 |
| 151 | G | - | - | P | 217 | 0.608714 |
| 152 | G | - | - | G | 218 | 0.433156 |
| 153 | G | - | - | A | 219 | 0.529335 |
| 154 | H | - | - | A | 220 | 0.347968 |
| 155 | H | - | - | I | 221 | 0.28936 |
The match states are 0-indexed in this dataframe.
And so with this class, anvi’o has easy access to IC for each match state in an .hmm file. One
need only initiate the above class as demonstrated.
The distribution of information content
Rather than testing for information content (IC) > 4, I wanted to keep the threshold a tunable parameter for the user (with a default being 4). To help guide anyone’s investigation, I have compiled all of the IC values for each match state in the representable InteracDome dataset (2375 HMM profiles). This totals 414619 IC values. Here is a histogram of them:

Wow, an absolutely gorgeous long-tailed distribution exhibiting Pareto-like qualities. Here it is in log-log:
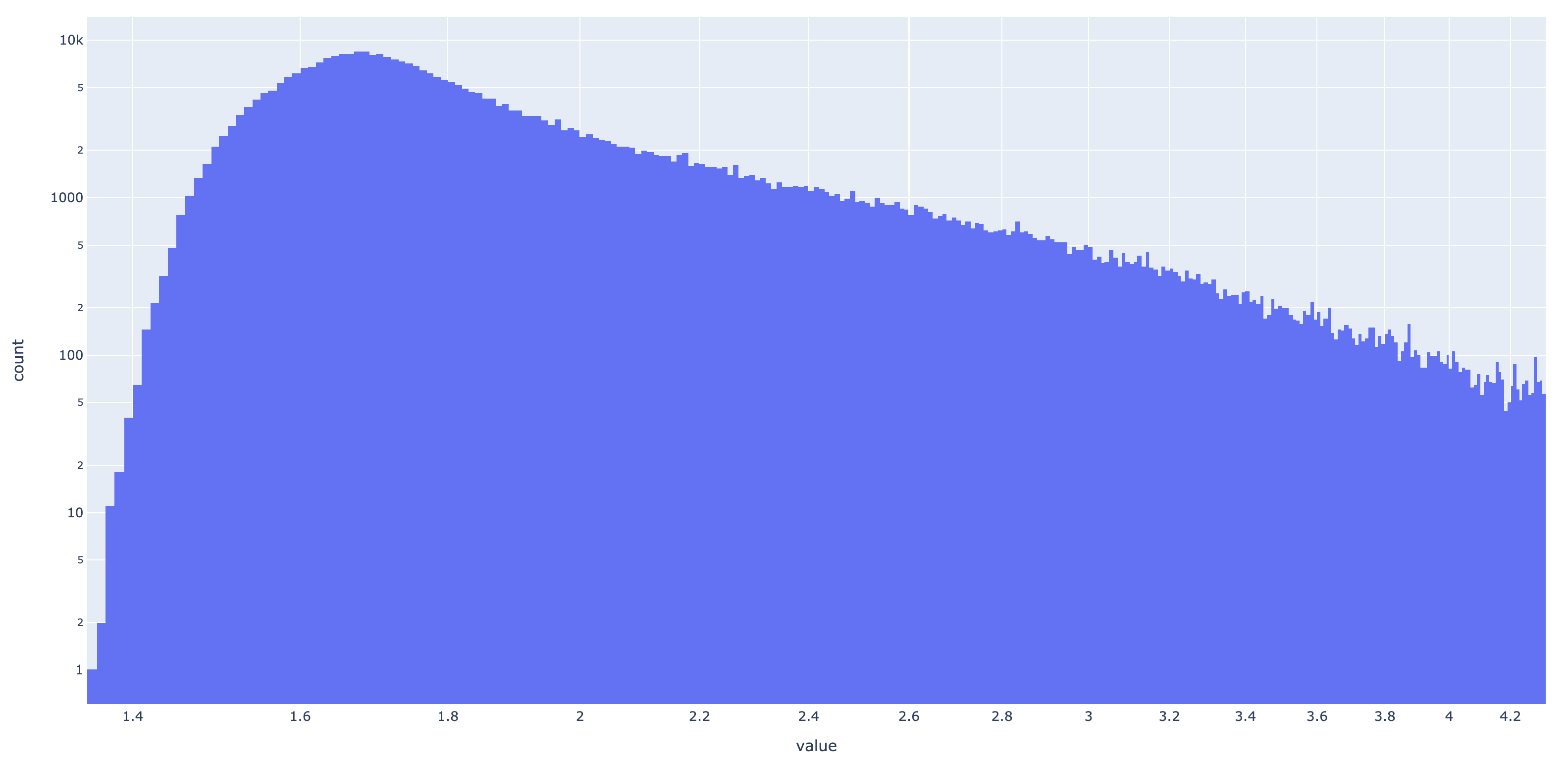
I digress–it’s shape does not so much matter. But what I do find interesting is that only a very small portion of sites have IC > 4, which is the cutoff threshold Kobren and Singh applied. Only 0.062% of match states have IC > 4, and on average, each HMM profile contains only 0.11 such match states. To see the effect of applying this filter to real hits, I did a series of tests with decreasing cutoff thresholds of IC:
| total hits | IC cutoff | filtered |
|---|---|---|
| 2898 | 4 | 0 |
| 2898 | 3.5 | 30 |
| 2898 | 3.0 | 151 |
| 2898 | 2.5 | 561 |
| 2898 | 2.0 | 1380 |
| 2898 | 1.5 | 2071 |
It is impossible to really say whether decreasing the IC cutoff below 4 is a good or bad idea without taking a close examination of hits that are filtered. I did not do this. What I do agree with is that IC > 4 match states are extremely rare, and thus represent the pinnacle of what should be conserved an ultra-conserved residue. If the sequence mismatches to the consensus value at these positions, we should definitely be justified in throwing the hit out. It is at this moment unclear if we could justifiably lower this threshold, or whether that would be throwing away useful data.
Cysteine & glycine are stubborn
Fun fact, this table shows the consensus of match states with IC > 4, partitioned by amino acid.
| AA | Number |
|---|---|
| C | 115 |
| G | 79 |
| P | 16 |
| D | 15 |
| H | 15 |
| R | 4 |
| W | 4 |
| K | 2 |
| N | 2 |
| E | 2 |
| Q | 1 |
| A | 1 |
| T | 1 |
Cyteine and glycine are astonishingly more likely to be ultra-conserved than any other residue.
(5) Matching binding frequencies to the genes’ residues
The flow
Once domain hits have been filtered, we are ready to attribute binding frequencies. The information flow of binding frequencies looks like this:
[InteracDome] [HMMER]
(binding frequency) ------------> (match state) ------> (user gene residue)
Binding frequencies are stored in either the conserved or representable InteracDome table and are attributed to match states of the IPfams. This is a mapping of binding frequency to match state. By running HMMER, we extracted alignment information of match states to user genes. This creates a mapping between user gene residues and match states, and by extension a mapping between user gene residues and binding frequencies. Because of the legwork already described in (4), there is very little to describe.
The most noteworthy thing to talk about here is how I dealt with multiple overlapping hits.
Dealing with multiple overlapping hits
Binding frequencies are held in a dataframe like this:
| gene_callers_id | codon_order_in_gene | pfam_id | match_state | ligand | binding_freq |
|---|---|---|---|---|---|
| 1 | 169 | PF00534 | 22 | ADP | 0.687948 |
| 1 | 169 | PF13692 | 8 | ADP | 0.595441 |
| 1 | 174 | PF00534 | 27 | ADP | 0.735759 |
| 1 | 174 | PF13692 | 14 | ADP | 0.595441 |
| 1 | 184 | PF00534 | 37 | ADP | 0.0697656 |
| 1 | 184 | PF13692 | 24 | ADP | 0.101399 |
| 1 | 186 | PF00534 | 39 | ADP | 0.0697656 |
| 1 | 186 | PF13692 | 26 | ADP | 0.101399 |
| 1 | 187 | PF13692 | 27 | ADP | 0.201761 |
| 1 | 189 | PF00534 | 47 | ADP | 0.0697656 |
This is a great format, because not only do you have binding frequencies associated with the
residues of user gene sequences (codon_order_in_gene), you also can see the exact match states
that contributed the binding frequency. But this has redundant entries. What I want is a single
binding frequency (for a given ligand) associated to each codon_order_in_gene, yet this format
allows redundancies. For example, further down the table there are these two entries:
| gene_callers_id | codon_order_in_gene | pfam_id | match_state | ligand | binding_freq |
|---|---|---|---|---|---|
| 1 | 167 | PF00534 | 20 | ALL_ | 0.0979682 |
| 1 | 167 | PF13692 | 6 | ALL_ | 0.103877 |
Here, we have an amino acid position that has two contributions, both from match_state 20 of
pfam_id PF00534, and match_state 6 of pfam_id PF13692. To condense this information, I created
a second dataframe that collapses the redundant rows. For simplicity, I am quite simply averaging
all binding frequencies (for a given ligand) that associate to a given amino acid position. The
above table is stored in the bind_freq attribute of anvio.interacdome.InteracDomeSuper, and the
second, collapsed table is stored in the avg_bind_freq attribute of the same class. In the redundant
case above, the corresponding entry in avg_bind_freq looks like this:
| gene_callers_id | ligand | codon_order_in_gene | binding_freq |
|---|---|---|---|
| 1 | ALL_ | 167 | 0.100922 |
match_state and pfam_id are dropped since these are no longer meaningful columns. However, their
information is still held in bind_freq (and as you will see in the next section, bind_freq is
stored as a tab-separated file so this potentially very useful information is not lost to the user).
Filtering low binding frequency scores
If users are not interested in positions with low binding frequency scores, they can apply a filter
with --min-binding-frequency and all entries below this threshold will be removed. This is also a
very convenient way to maintain reasonable table size for storage in the contigs database. Here is a
histogram of non-zero binding frequencies from a test dataset:
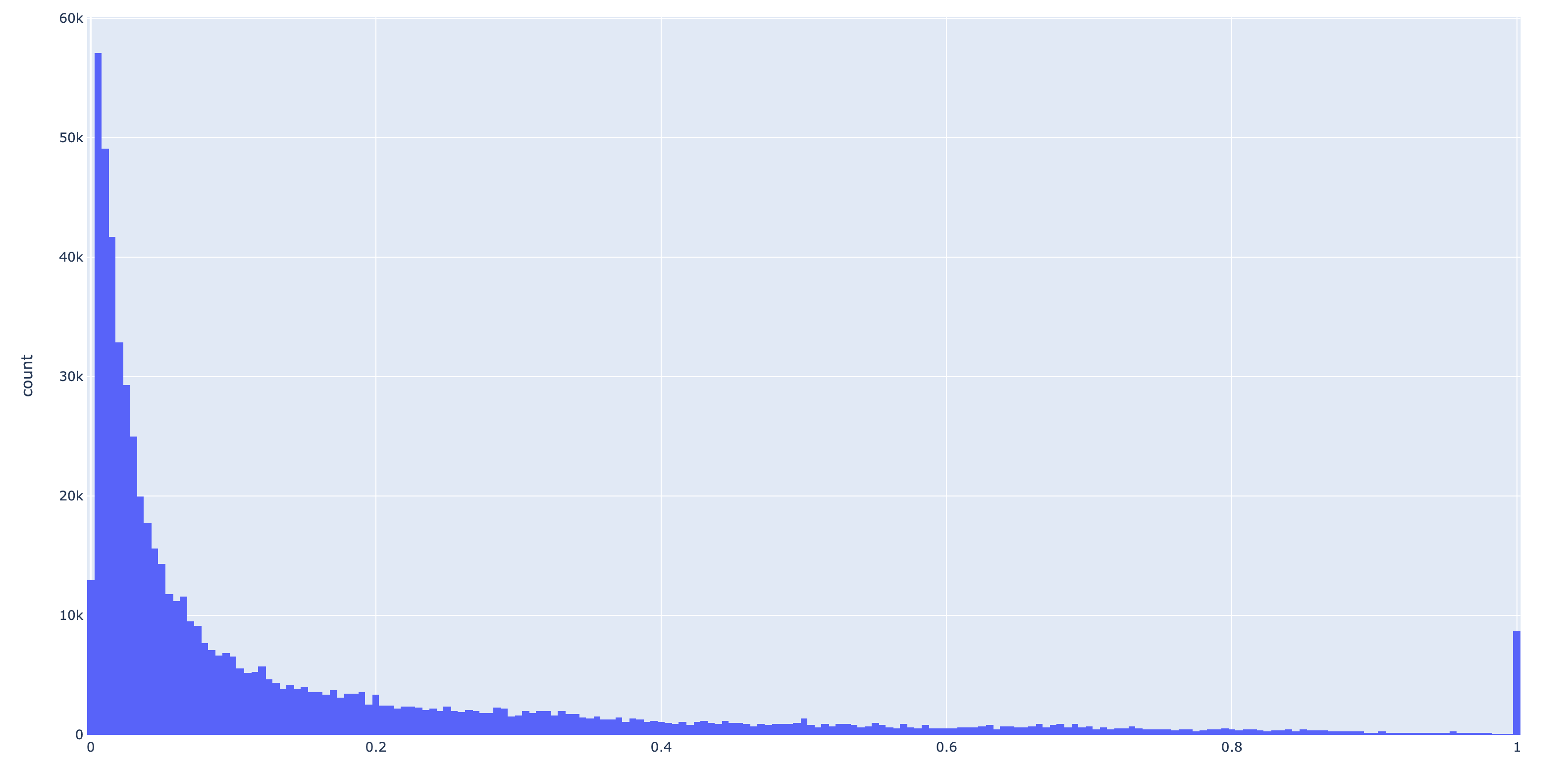
It’s clear from the above figure that even if the user is comfortable with discarding frequencies < 0.05, it has huge effect on table size.
(6) Storing the per-residue binding frequencies into the contigs database
The data stored in the contigs database is a variant of avg_bind_freq. As a reminder,
here is what avg_bind_freq looks like this:
| gene_callers_id | ligand | codon_order_in_gene | binding_freq |
|---|---|---|---|
| 1 | ADP | 169 | 0.641695 |
| 1 | ADP | 174 | 0.6656 |
| 1 | ADP | 184 | 0.0855824 |
| 1 | ADP | 186 | 0.0855824 |
| 1 | ADP | 187 | 0.201761 |
| 1 | ADP | 189 | 0.0855824 |
| 1 | ADP | 190 | 0.163235 |
| 1 | ADP | 192 | 0.095622 |
| 1 | ADP | 196 | 0.262405 |
| 1 | ADP | 199 | 0.161423 |
And here is the corresponding section of what is ultimately stored in the contigs database:
| item_name | data_key | data_value | data_type | data_group |
|---|---|---|---|---|
| 1:169 | ADP | 0.641695 | float | InteracDome |
| 1:174 | ADP | 0.6656 | float | InteracDome |
| 1:184 | ADP | 0.0855824 | float | InteracDome |
| 1:186 | ADP | 0.0855824 | float | InteracDome |
| 1:187 | ADP | 0.201761 | float | InteracDome |
| 1:189 | ADP | 0.0855824 | float | InteracDome |
| 1:190 | ADP | 0.163235 | float | InteracDome |
| 1:192 | ADP | 0.095622 | float | InteracDome |
| 1:196 | ADP | 0.262405 | float | InteracDome |
| 1:199 | ADP | 0.161423 | float | InteracDome |
It may seem to be in oddly specific format, and that’s because it is. You see, up until a few months
ago, anvi’o contigs databases had no means of storing per-nucleotide or per-residue information.
Crazy, I know. The only thing that resembled this concept was per-nucleotide coverage values, but
this is actually stored in the auxiliary database (a tumor of the profile database) that’s created
during anvi-profile. A few months ago I wanted to address this shortcoming for the selfish reason
of knowing that one day I would implement InteracDome functionality into anvi’o. And I didn’t want
anvi’o to spit out some half-chewed tab-separated file to the user–I wanted the results to be
stored in the contigs database so they could be used in integrated and interactive manners. So I created two
new tables in the contigs database called amino_acid_additional_data and
nucleotide_additional_data, which hijack the already existing framework for additional data
tables. The convenience factor was through the
roof for doing this, as I had to write very little code (here is the
pull request) and now we can store arbitrary per-nucleotide and per-amino-acid annotations (not just
InteracDome stuff, but practically anything besides .mp4 format). However, two downsides are very appreciable.
First, the framework requires a unique key for each amino acid. Yet, it is technically two keys that
define an amino acid: the gene_callers_id of the gene it belongs to, and the codon_order_in_gene
(the residue position) of the amino acid relative to its gene. The same goes for defining a nucleotide: you need the position
in the contig (pos_in_contig) and you need the contig_name. So how do you get one key from two
pieces of information? The solution, which is ugly, is to create a single key by concatenating these
two pieces of information into a string. Doing so has distinct disadvantages, the main one being that
SQL queries are hindered: it becomes impossible to straight-forwardly grab all amino acids belonging to a gene without first
loading all entries into memory, for example. Regardless, I did it. Someone will hate me in 5 years.
Take another look at the above table and you will see the string concatenation I’m talking about under the item_name
column. Entries under item_name equal to <gene_callers_id>:<codon_order_in_gene>, for example the first
entry 1:169 is the 169th residue (0-indexed) in gene 1.
The second downside to doing this is specific to anvi-run-interacdome. You see, there is really a lot of
information missing from the above table. For example, it would be great to know which IPfam ID(s)
contributed a given binding freqency and with which of their match states they did so. I already discussed that
this information is all present in the dataframe bind_freq. Unfortunately there just isn’t
enough columns to accomodate this information, and I am fixed within this framework because I didn’t want to reinvent the wheel. To
console victims of this tyranny, anvi-run-interacdome does more than just store the above table in
the contigs database. It also outputs two tab-delimited files. Their names are by default
INTERACDOME-match_state_contributors.txt and INTERACDOME-domain_hits.txt…
INTERACDOME-match_state_contributors.txt is a table of the exact contents in bind_freq
mentioned above. Since you’re somehow still reading
this, I’ll show you the table again:
| gene_callers_id | codon_order_in_gene | pfam_id | match_state | ligand | binding_freq |
|---|---|---|---|---|---|
| 1 | 169 | PF00534 | 22 | ADP | 0.687948 |
| 1 | 169 | PF13692 | 8 | ADP | 0.595441 |
| 1 | 174 | PF00534 | 27 | ADP | 0.735759 |
| 1 | 174 | PF13692 | 14 | ADP | 0.595441 |
| 1 | 184 | PF00534 | 37 | ADP | 0.0697656 |
| 1 | 184 | PF13692 | 24 | ADP | 0.101399 |
| 1 | 186 | PF00534 | 39 | ADP | 0.0697656 |
| 1 | 186 | PF13692 | 26 | ADP | 0.101399 |
| 1 | 187 | PF13692 | 27 | ADP | 0.201761 |
| 1 | 189 | PF00534 | 47 | ADP | 0.0697656 |
From this, one can trace back each and every match state that contributed to a given binding frequency. This is what giving power to the user means.
INTERACDOME-domain_hits.txt is a table of the exact contents in dom_hits
mentioned above. Since you’re somehow still reading
this, I’ll show you the table again:
| pfam_name | pfam_id | corresponding_gene_call | domain | qual | score | bias | c-evalue | i-evalue | hmm_start | hmm_stop | hmm_bounds | ali_start | ali_stop | ali_bounds | env_start | env_stop | env_bounds | mean_post_prob | match_state_align | comparison_align | sequence_align | version |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta_elim_lyase | PF01212 | 1762 | 1 | ! | 20.9 | 0.1 | 1e-08 | 3.5e-06 | 33 | 169 | .. | 44 | 177 | .. | 34 | 215 | .. | 0.72 | tvnrLedavaelfgke..aalfvpqGtaAnsill.kill.qr..geevivtepahihfdetgaiaelagvklrdlknkeaGkmdlekleaaikevgaheekiklisltvTnntagGqvvsleelrevaaiakkygiplhlDgA | ++ +++ael+ + f+ Gt +++ l + + +r g+ +i++ h +et + g +l ++ +++G +++e+l+++i++ e i + +++v n+ G++ +++e+ ev +a+ +i++h+D+ | LLQQARKQIAELINVSanEIYFTSGGTEGDNWVLkGTAIeKRefGNHIIISAVEHPAVTETAEQLVELGFELSYAPVDKEGRVKVEELQKLIRK—–ETILVSVMAVNNE–VGTIQPIKEISEV–LAEFPKIHFHVDAV | 20 |
| PAPS_reduct | PF01507 | 1541 | 1 | ! | 36.1 | 0.1 | 3.6e-13 | 1.3e-10 | 2 | 164 | .. | 21 | 231 | .. | 20 | 234 | .. | 0.79 | lvvsvsgGkdslVllhLalkafkpv….pvvfvdtghefpetiefvdeleeryglrlkvyepeeevaekinaekhgs.slyee.aaeriaKveplkk……………………………aLekldedall..tGaRrdesksraklpiveidedfek………slrvfPllnWteedvwqyilrenipynpLydqgfr | + +s+sgGkds +++La + ++ ++ ++ + ++ t++f++++e+ +++ +++ ++++ + + +++ + + + e+ + p k e++ ++a+ +G+R++es +r++ +++ +++ + ++Pl++W+ d+w+ + +++yn +y++ ++ | VYFSFSGGKDSGLMVQLANLVAEKLdrnfDLLILNIEANYTATVDFIKKIEQLPRVKNIYHFCLPFFEDNNTSFFQPQwKMWDPsEKEKWIHSLP–KnaitleniddglkkyyslsngnpdrflryfqnwYKEQYPQSAIScgVGIRAQESLHRHSAVTKGENKYKNRcwinitlegNILFYPLFDWKVGDIWAATFKCELEYNYIYEKMYK | 18 |
| Ank_2 | PF12796 | 1756 | 1 | ! | 32.2 | 0 | 6.7e-12 | 2.3e-09 | 29 | 84 | .] | 74 | 135 | .. | 53 | 135 | .. | 0.85 | aLhyAakngnleivklLle…h.a..adndgrtpLhyAarsghleivklLlekgadinlkd | aL Aa + +++ vk +l+ + + +d +g+tpL +A+ ++ +ei+k L+++gadinl++ | ALLEAANQRDTKKVKEILQdttYqVdeVDTEGNTPLNIAVHNNDIEIAKALIDRGADINLQN | 6 |
| Ank_2 | PF12796 | 1756 | 2 | ! | 28.5 | 0 | 9.5e-11 | 3.3e-08 | 22 | 75 | .. | 199 | 265 | .. | 195 | 267 | .] | 0.76 | pn..k.ngktaLhyAak..ngnl…eivklLleha…..adndgrtpLhyAarsghleivklLle | ++ + +g taL+ A+ +gn +ivklL+e++ dn+grt++ yA ++g++ei k+L + | IDfqNdFGYTALIEAVGlrEGNQlyqDIVKLLMENGadqsiKDNSGRTAMDYANQKGYTEISKILAQ | 6 |
| IGPS | PF00218 | 1615 | 1 | ! | 20.6 | 0.1 | 1.2e-08 | 4e-06 | 202 | 249 | .. | 195 | 242 | .. | 73 | 248 | .. | 0.88 | LaklvpkdvllvaeSGiktredveklkeegvnafLvGeslmrqedvek | +++lv+++++++ae i+t+e+++++k+ gv ++ vG +++r ++ +k | IKQLVQENICVIAEGKIHTPEQARQIKKLGVAGIVVGGAITRPQEIAK | 20 |
| Ribosomal_L33 | PF00471 | 1562 | 1 | ! | 66.6 | 1.5 | 1.1e-22 | 3.7e-20 | 2 | 47 | .] | 4 | 49 | .] | 3 | 49 | .] | 0.97 | kvtLeCteCksrnYtttknkrntperLelkKYcprcrkhtlhkEtK | +++LeC e+++r Y t+knkrn+perLelkKY p++r++ ++kE K | NIILECVETGERLYLTSKNKRNNPERLELKKYSPKLRRRAIFKEVK | 19 |
| Ribosomal_S14 | PF00253 | 1565 | 1 | ! | 83.3 | 0.1 | 3.9e-28 | 1.3e-25 | 2 | 54 | .] | 36 | 88 | .. | 35 | 88 | .. | 0.98 | laklprnssptrirnrCrvtGrprGvirkfgLsRicfRelAlkgelpGvkKaS | laklpr+s+p+r+r r++ +GrprG++rkfg+sRi+fRel ++g +pGvkKaS | LAKLPRDSNPNRLRLRDQTDGRPRGYMRKFGMSRIKFRELDHQGLIPGVKKAS | 20 |
| Polysacc_synt_C | PF14667 | 1593 | 1 | ! | 61.4 | 19.2 | 5.4e-21 | 1.9e-18 | 2 | 139 | .. | 371 | 516 | .. | 370 | 519 | .. | 0.83 | LailalsiiflslstvlssiLqglgrqkialkalvigalvklilnllliplfgivGaaiatvlallvvavlnlyalrrllgikl…llrrllkpllaalvmgivvylllllllglllla…al..alllavlvgalvYllllll | L+ ++s+ +l+++t++ siLq+l +k+a+ ++ i++l+kli+++++i+lf +G +iat+++ ++++++ +++l+r++ i++ ++ +++ +++vm i+ +l+l+++ ++ + +l + l +++g++v+ + l++ | LSATIISTSLLGIFTIVLSILQALSFHKKAMQITSITLLLKLIIQIPCIYLFKGYGLSIATIICTMFTTIIAYRFLSRKFDINPikyNRKYYSRLVYSTIVMTILSLLMLKIISSVYKFEstlQLffLISLIGCLGGVVFSVTLFR | 5 |
From this, one can learn about how good a given hit was, the alignment of the user gene to the HMM match states, and hopefully anything else the user would be interested in.
Conclusion
Wow! That was certainly a mouthful. I know in a year or two when I am writing my thesis, this will be helpful to me, and I hope that it has been helpful to you too. If you have questions, let me know. And if you want to see all of this in action, be sure to check out the InteracDome section of the infant gut tutorial.