



Table of Contents
The anvi’o metagenomic workflow assumes that you have metagenomic short reads. But what if all you have is a bunch of contigs, or a draft genome, or a MAG without any short reads to map to?
This need was brought up by one of our early users, and there has been an open issue to address this at some point. It is now resolved, and the following functionality is available in the master branch.
The key is to create a blank anvi’o profile database to go with the contigs database, and this is what I will demonstrate here. But before I start, let me put this here so you know what version I am using:
$ anvi-profile -v
Anvi'o version ...............................: 2.0.0
Profile DB version ...........................: 15
Contigs DB version ...........................: 5
Samples information DB version ...............: 2
Auxiliary HDF5 DB version ....................: 1
Users DB version (for anvi-server) ...........: 1
Preparing the FASTA file
For this example I will download a this Bacillus subtilis genome, which was sequenced by Mitsuhiro Itaya et al., and was made available to the community along with their publication with an awesome title:
“Combining two genomes in one cell: Stable cloning of the Synechocystis PCC6803 genome in the Bacillus subtilis 168 genome”
OK. The reason I will use this genome is simple: it is a chimeric genome, and it will be fun to see that visually.
This is how I downloaded the genome:
$ wget ftp://ftp.ensemblgenomes.org/pub/current/bacteria/fasta/bacteria_25_collection/bacillus_subtilis_best7613/dna/Bacillus_subtilis_best7613.ASM32874v1.31.dna.genome.fa.gz -O Bacillus_subtilis.fa.gz
$ gzip -d Bacillus_subtilis.fa.gz
$ ls -lh Bacillus_subtilis.fa
-rw-r--r-- 7.4M Jun 6 14:42 Bacillus_subtilis.fa
Simple. As you may know, anvi’o requires simple deflines in FASTA files), and this FASTA file does not conform that requiremenet at all:
$ head -n 1 Bacillus_subtilis.fa
>Chromosome dna:chromosome chromosome:ASM32874v1:Chromosome:1:7585470:1
So I need to fix that first before I generate the contigs database. For this purpose I use anvi-script-remove-short-contigs-from-fasta with a minimum length of 0 and --simplify flag. This way the script will not remove anything from the FASTA file while converting deflines to simpler ones:
$ anvi-script-remove-short-contigs-from-fasta Bacillus_subtilis.fa --min-len 0 --simplify-names -o fixed.fa
Input ........................................: Bacillus_subtilis.fa
Output .......................................: fixed.fa
Minimum length ...............................: 0
Total num contigs ............................: 1
Total num nucleotides ........................: 7,585,470
Contigs removed ..............................: 0 (0.00% of all)
Nucleotides removed ..........................: 0 (0.00% of all)
Deflines simplified ..........................: True
$ mv fixed.fa Bacillus_subtilis.fa
Now it looks much better:
$ head -n 1 Bacillus_subtilis.fa
>c_000000000001
So this is my FASTA file, and you have your’s. We are golden. The next step is to create an anvi’o contigs database.
Creating the contigs database
Creating a contigs database is not much different for this workflow than any other. However, (1) I will set a shorter split size than default (since I want to see a more highly resolved depiction of the genome), and (2) I will skip mindful splitting, so anvi’o does not lose time with something that is not useful in this context:
anvi-gen-contigs-database -f Bacillus_subtilis.fa \
-o Bacillus_subtilis.db \
-L 5000 \
--skip-mindful-splitting \
--name 'B. subtilis'
Next, I populate the single-copy gene hit tables in this newly generated contigs database:
anvi-run-hmms -c Bacillus_subtilis.db
If you hapenned to read this post, you already know that at this point we can take a look at the distribuiton of bacterial single-copy genes in this contigs database and predict the number of genomes in it:
Please use the program anvi-display-contigs-stats to learn about the predicted number of bacterial, archaeal, and eukaryotic genomes you have in your contigs database. The interactive display of that program will give you these numbers also. We are keeping the rest of the post here so we remember where we are coming from :)
$ anvi-script-gen_stats_for_single_copy_genes.py Bacillus_subtilis.db
$ anvi-script-gen_stats_for_single_copy_genes.R Bacillus_subtilis.db.hits Bacillus_subtilis.db.genes
Which gives me back this PDF image:
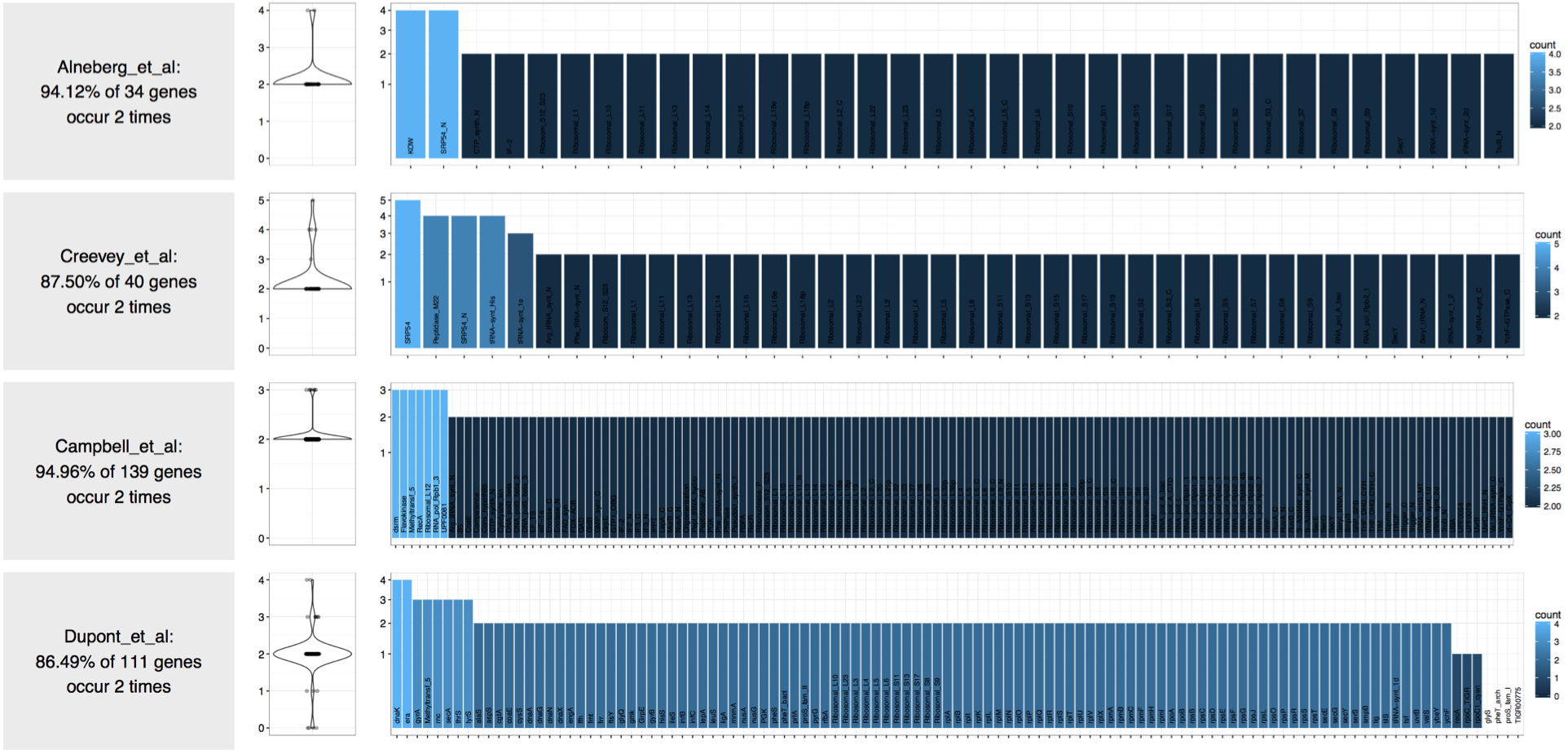
This output confirms that there are two genomes in this contigs database. This was not a case of contamination, but let’s assume it was. So you did things, and figured out that your draft genome is likely contamianted.
Learning that your genome (or your draft genome, or your MAG, you name it) is contaminated is always very important (because we know what happens when you never learn it and continue analyzing what you have), but the realization of contamination gives you nothing but a dark afternoon if you don’t know what is the best way to deal with it. When there is mapping data available, anvi’o offers powerful ways to characterize contamination as we recently demonstarted in our publication “Identifying contamination with advanced visualization and analysis practices”.
But none of that works if you don’t short reads to map, and the next step will enable us to bypass that need.
Creating a blank profile database
To visualize a contigs database, we need an anvi’o profile database. But what does one do if there is no mapping data to create a profile database? Well, they run anvi-profile with --blank-profile parameter!
$ anvi-profile -c Bacillus_subtilis.db -o Bacillus_subtilis -S Bacillus_subtilis --blank-profile
Thanks to the resulting blank profile database, now I will be able to visualize what this contigs database holds in it by using anvi-interactive. Now I can do binning with real time completion / contamination estimates, store and load states, create collections, and summarize them.
Visualizing
OK. Everything is ready to simply start the anvi’o interactive interface for a better look:
anvi-interactive -c Bacillus_subtilis.db -p Bacillus_subtilis/PROFILE.db
Which gives me this view in the opening window:
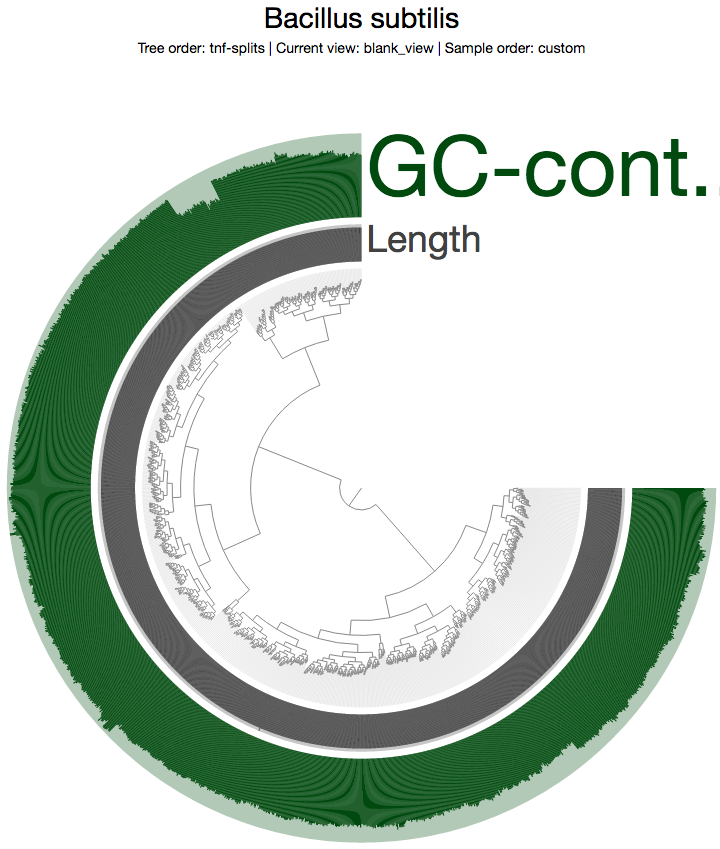
The inner tree shows the organization of each 5 kbp split in this genome based on their tetra-nucleotide frequency profiles. As you can see, there are two distinct branches in the tree. One of these branches probably represents the Synechocystis genome, and the other represents the Bacillus subtilis genome Mitsuhiro Itaya et al. masterfully merged together.
Although this is the simplest look at this data for the sake of clarity, you can indeed use --additional-view, or --additional-layers to add more data into the interface, and do pretty much anything you would do with any other anvi’o project.
As an example, here I will separate those genomes from each other by selecting splits represented in those distinct branches into different bins, and then store my selections into a ‘collection’ to summarize it later (click to load the animated gif):
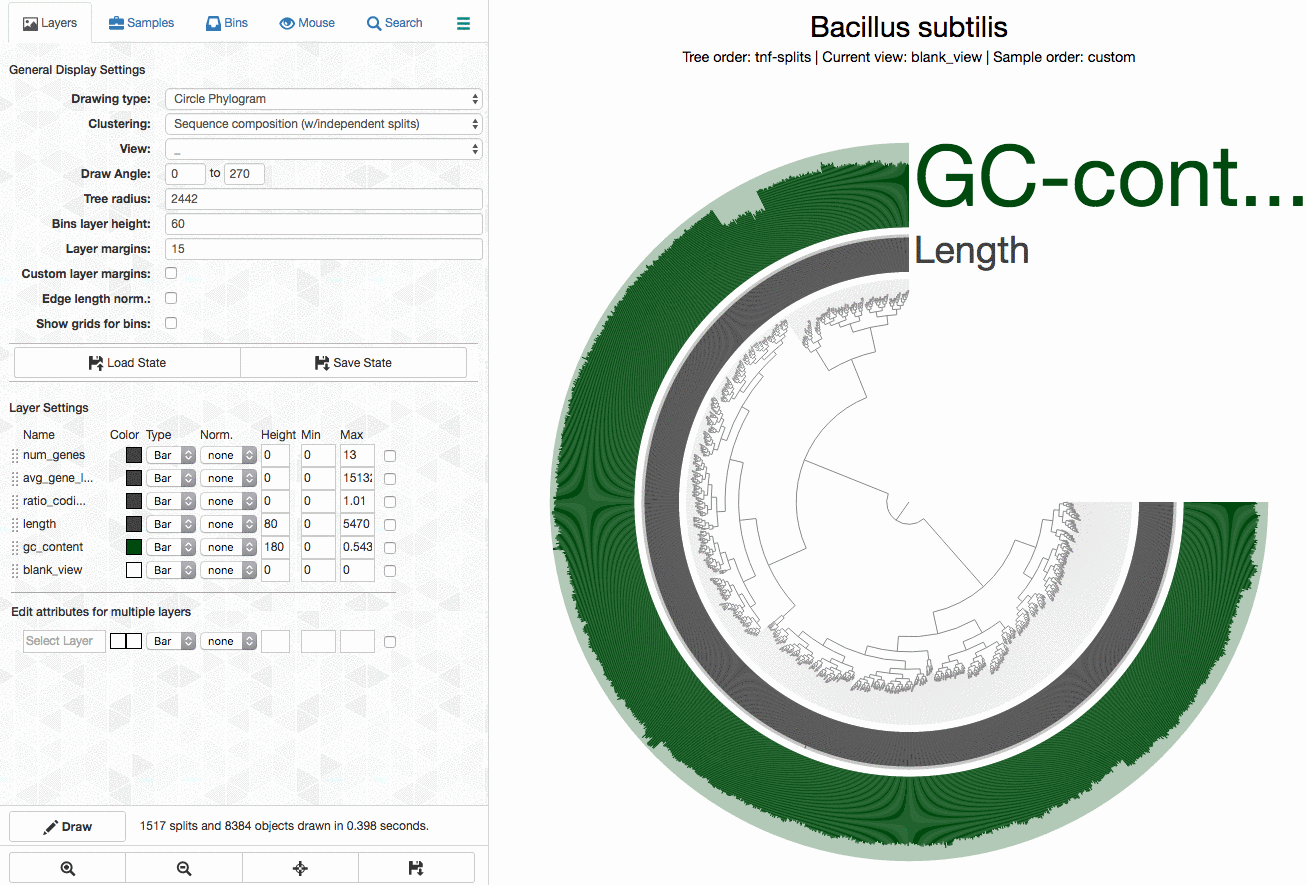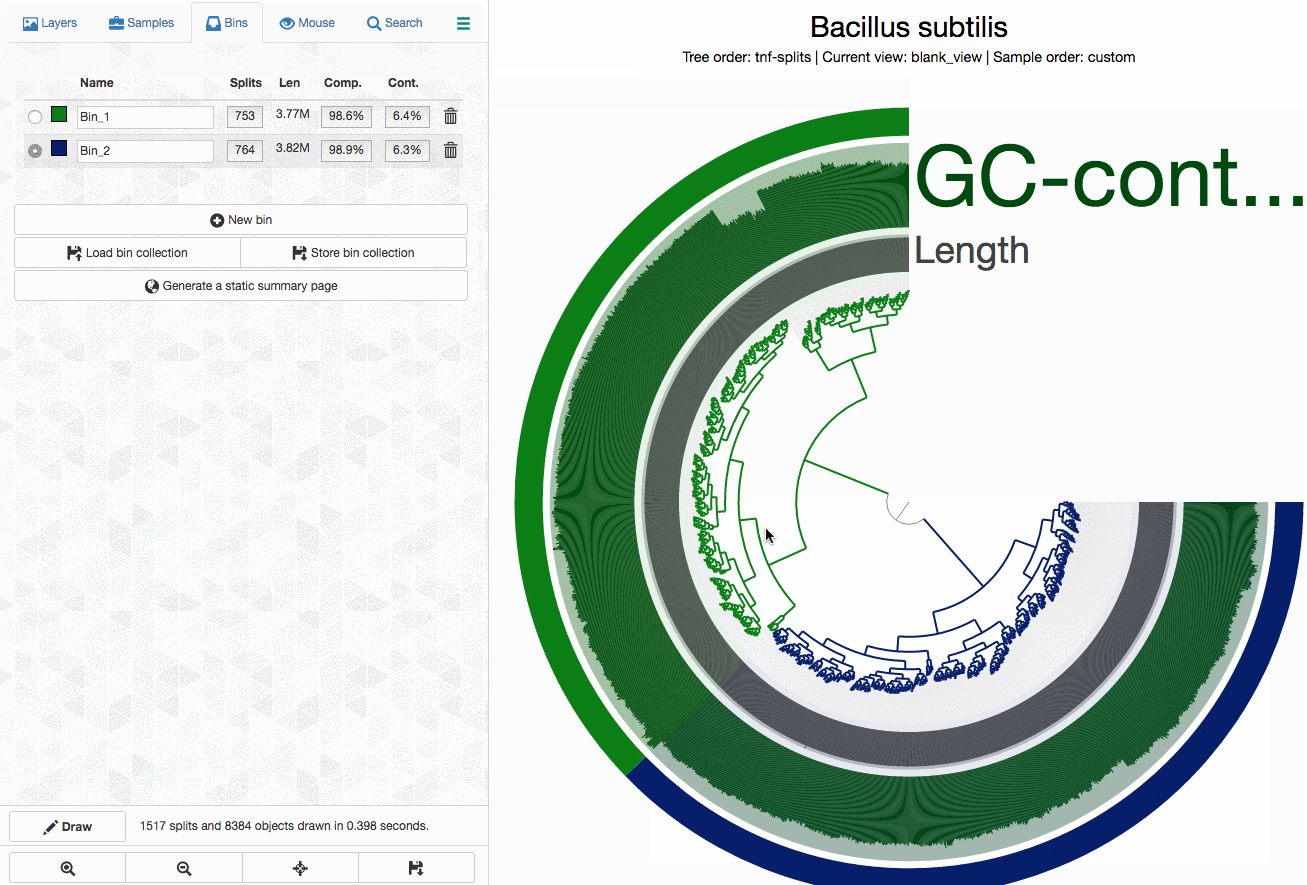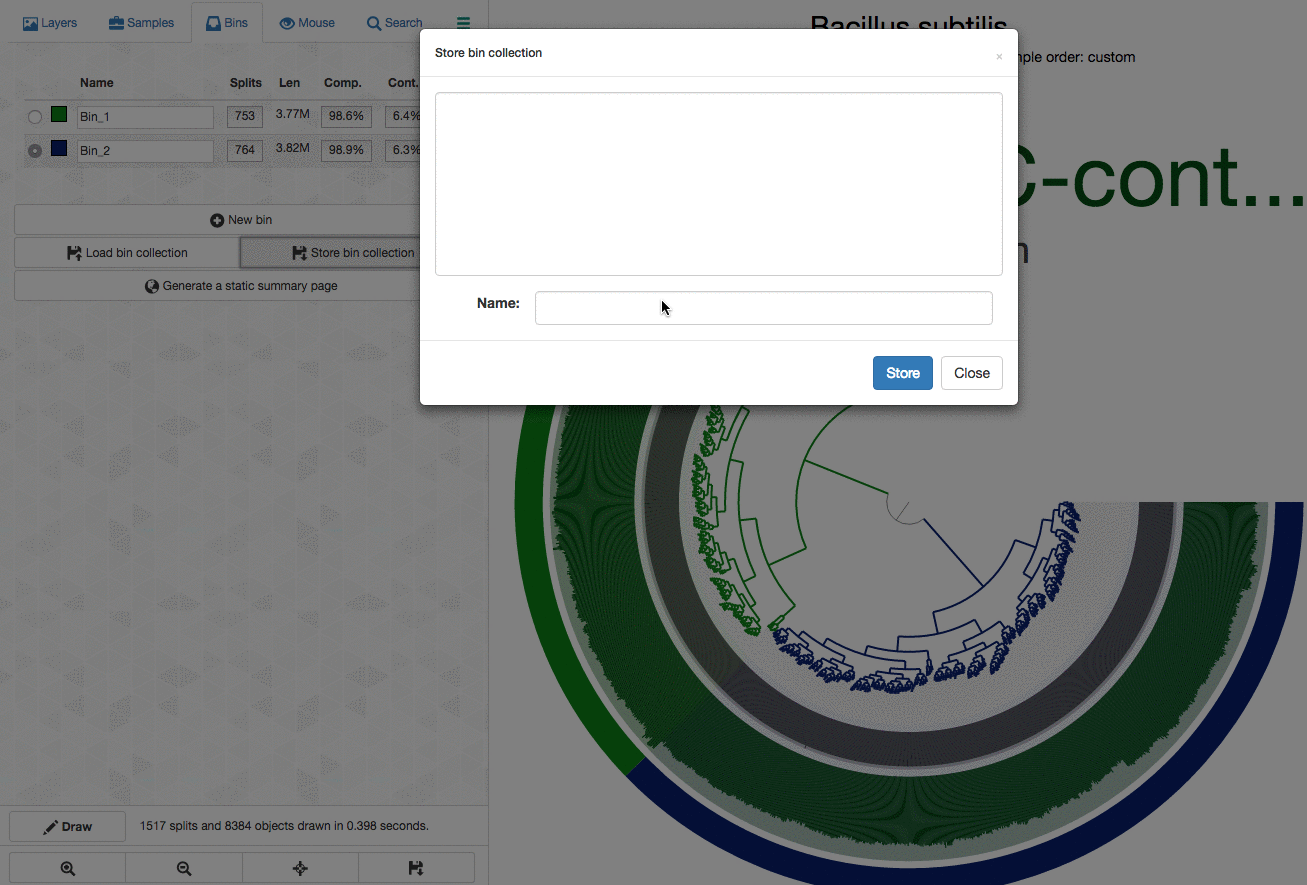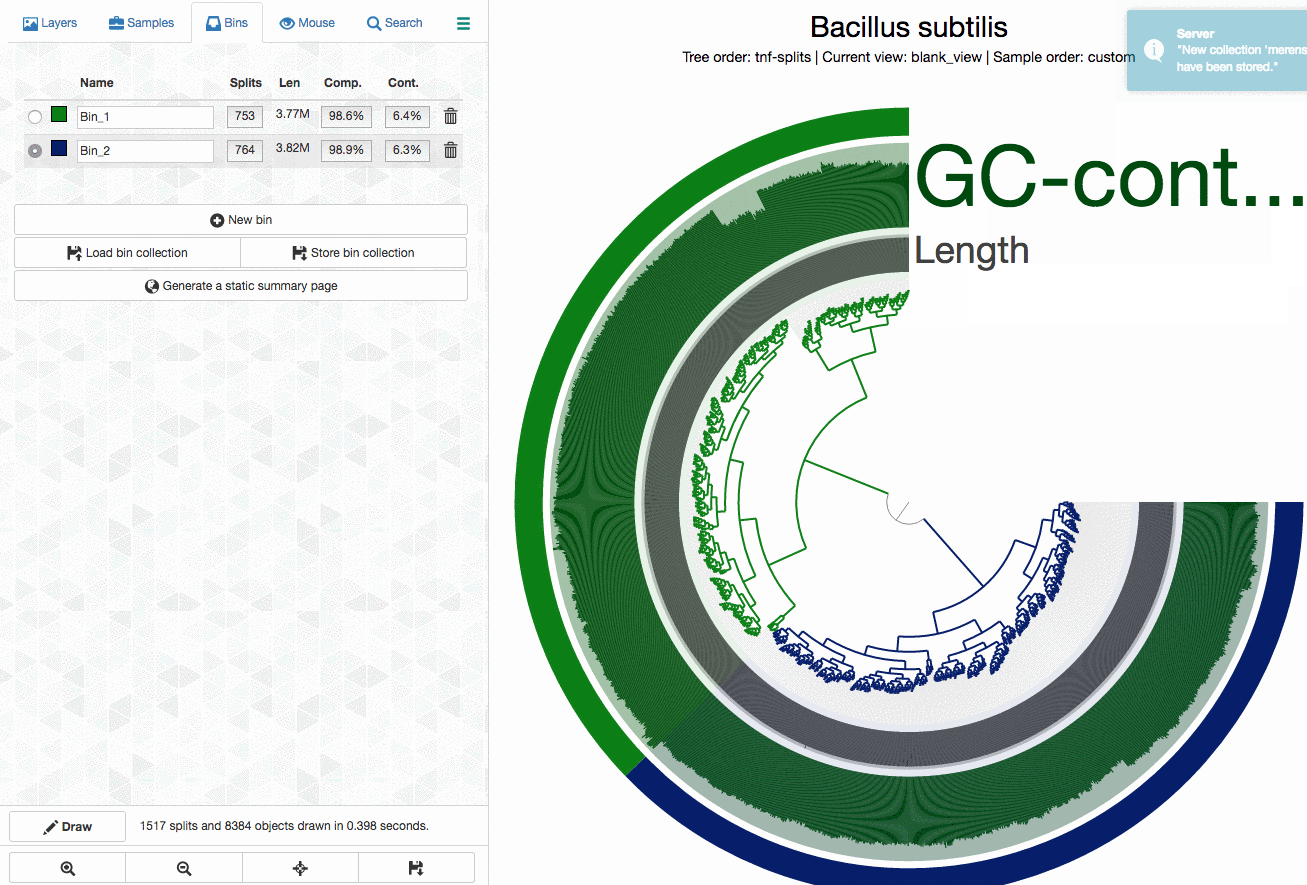
As you can see, completion / redundancy estimates look much better when they are selected separately.
Summary
Now I can summarize the collection I stored in the blank profile database:
anvi-summarize -c Bacillus_subtilis.db -p Bacillus_subtilis/PROFILE.db -C merens -o Bacillus_subtilis_summary
The result of this summary is a static HTML output. I copied that directory here for your viewing pleasure:
http://anvio.org/data/Bacillus_subtilis_summary/
Mission accomplished!