



Table of Contents
The latest version of anvi’o is v7.1. See the release notes.
This feature is available in anvi’o v2.3.3 or later. You can learn which version you have on your computer by typing anvi-profile --version in your terminal.
TL;DR
In most cases, getting ribosomal RNA genes out of isolate or metagneome-assembled genomes is not as straightforward as one would like. Gene callers usually don’t perform well when it comes to identifying 16S or 23S rRNA genes, and using primer sequences to find these regions in genomes is too 2005. Luckily in anvi’o you can accomplish exactly that using these three anvi’o programs that work in tandem: anvi-gen-contigs-database, anvi-run-hmms, and anvi-get-sequences-for-hmm-hits. For any given FASTA file, it will work like this:
anvi-gen-contigs-database -f GENOME.fa -o GENOME.db
anvi-run-hmms -c GENOME.db
anvi-get-sequences-for-hmm-hits -c GENOME.db --hmm-source Ribosomal_RNA_16S
So you can spend more time on the next thing on your bottomless TODO list.
Please note that you can use the program anvi-db-info on any contigs-db to learn about different “HMM sources” to go with the --hmm-source parameter.
TL
We always wanted to be able to show rRNA genes in our anvi’o displays.
Although genes with many conserved regions such as the 16S rRNA gene often fail to remain in our assemblies, some of our bins did contain these genes occasionally. In those cases, while you work with your bins, of course it would have been lovely to see contigs that happen to contain rRNA genes, and maybe get those sequences and quickly BLAST them, etc.
Thanks to Torsten Seemann’s previous efforts for Barrnap, we now have an operational HMM collection for rRNA genes (not perfect, but muuuuch better than nothing).
You will find in this post how to work with the HMM profile Ribosomal RNAs in anvi’o to get rRNA genes from a single FASTA file, or from anvi’o projects. There is also some technical blurb at the end of the post if you have too much time in your hands.
Getting rRNA gene hits from a FASTA file
Let’s assume you have a FASTA file. I will download a B. fragilis genome from European Nucleotide Archive just to give a reproducible example, and generate a contigs database for it:
# download the genome
wget "http://www.ebi.ac.uk/ena/data/view/CR626927.1,CR626928.1&download=fasta&display=fasta" -O B_fragilis_ATCC_25285.fa
# clean up the crappy defline
anvi-script-reformat-fasta B_fragilis_ATCC_25285.fa \
--simplify-names \
-o B_fragilis.fa
# create an anvi'o contigs database
anvi-gen-contigs-database -f B_fragilis.fa \
-o B_fragilis.db
Fine. Now we have a contigs database. The next step is to run HMM profiles, which, if you are using anvi’o v2.3.3 or later, will include the profile for ribosomal RNAs:
$ anvi-run-hmms -c B_fragilis.db --num-threads 10
Done! Now you can do all sorts of fancy stuff. Let’s get the 16S rRNA gene sequences:
# listing all HMMs in the contigs database
$ anvi-get-sequences-for-hmm-hits -c B_fragilis.db \
--list-hmm-sources
AVAILABLE HMM SOURCES
===============================================
* 'Archaea_76' (76 models with 70 hits)
* 'Bacteria_71' (71 models with 147 hits)
* 'Protista_83' (83 models with 116 hits)
* 'Ribosomal_RNA_12S' (1 model with 0 hits)
* 'Ribosomal_RNA_16S' (3 models with 7 hits)
* 'Ribosomal_RNA_18S' (1 model with 0 hits)
* 'Ribosomal_RNA_23S' (2 models with 7 hits)
* 'Ribosomal_RNA_28S' (1 model with 0 hits)
* 'Ribosomal_RNA_5S' (5 models with 0 hits)
# getting back sequences for 16S rRNA genes
$ anvi-get-sequences-for-hmm-hits -c B_fragilis.db \
--hmm-source Ribosomal_RNA_16S \
-o sequences.fa
Contigs DB ...................................: Initialized: B_fragilis.db (v. 20)
Sources ......................................: Ribosomal_RNA_16S
Hits .........................................: 2 HMM hits for 1 source(s)
Genes of interest ............................: None
Mode .........................................: DNA sequences
Genes are concatenated .......................: False
Output .......................................: sequences.fa
So far so good. One of the sequences in the file is this:
>16S_rRNA_arc___Ribosomal_RNA_16S___299d9f bin_id:B_fragilis|source:Ribosomal_RNA_16S|e_value:1.3e-239|contig:c_000000000001|gene_callers_id:10291|start:3137169|stop:3138643|length:1474
AGGAGGTGTTCCAGCCGCACCTTCCGGTACGGCTACCTTGTTACGACTTAGCCCCAGTCACCAGTTTTACCCTAGGACGATCCTTGCGGTTACGTACTTCAGGTACCCCCGGCTCCCATG
GCTTGACGGGCGGTGTGTACAAGGCCCGGGAACGTATTCACCGCGCCGTGGCTGATGCGCGATTACTAGCGAATCCAGCTTCACGAAGTCGGGTTGCAGACTTCGATCCGAACTGAGAGA
GGATTTTGGGATTAGCATACGGTCACCCGCTAGCTGCCTTCTGTACCCCCCATTGTAACACGTGTGTAGCCCCGGACGTAAGGGCCGTGCTGATTTGACGTCATCCCCACCTTCCTCACA
TCTTACGACGGCAGTCTCTCTAGAGTCCTCAGCATAACCTGTTAGTAACTAAAGATAAGGGTTGCGCTCGTTATGGCACTTAAGCCGACACCTCACGGCACGAGCTGACGACAACCATGC
AGCACCTTCACAGCGGTGATTGCTCACTGACATGTTTCCACATCATTCCACTGCAATTTAAGCCCGGGTAAGGTTCCTCGCGTATCATCGAATTAAACCACATGTTCCTCCGCTTGTGCG
GGCCCCCGTCAATTCCTTTGAGTTTCACCGTTGCCGGCGTACTCCCCAGGTGGAATACTTAATGCTTTCGCTTGGCCGCTTACTGTATATCGCAAACAGCGAGTATTCATCGTTTACTGT
GTGGACTACCAGGGTATCTAATCCTGTTTGATACCCACACTTTCGAGCATCAGTGTCAGTTGCAGTCCAGTGAGCTGCCTTCGCAATCGGAGTTCTTCGTGATATCTAAGCATTTCACCG
CTACACCACGAATTCCGCCCACCTCTACTGTACTCAAGACTGACAGTATCAACTGCAATTTTACGGTTGAGCCGCAAACTTTCACAACTGACTTACCAGTCCACCTACGCTCCCTTTAAA
CCCAATAAATCCGGATAACGCTCGGATCCTCCGTATTACCGCGGCTGCTGGCACGGAGTTAGCCGATCCTTATTCATATAATACATACAAAACAGTATACATACTGCACTTTATTCTTAT
ATAAAAGAAGTTTACGACCCATAGAGCCTTCATCCTTCACGCTACTTGGCTGGTTCAGGCTAGCGCCCATTGACCAATATTCCTCACTGCTGCCTCCCGTAGGAGTTTGGACCGTGTCTC
AGTTCCAATGTGGGGGACCTTCCTCTCAGAACCCCTATCCATCGAAGGCTTGGTGAGCCGTTACCTCACCAACAACCTAATGGAACGCATCCCCATCCTTTACCGGAATCCTTTAATAAT
GAAACCATGCGGAATCATTATGCTATCGGGTATTAATCTTTCTTTCGAAAGGCTATCCCCGAGTAAAGGGCAGGTTGGATACGTGTTACTCACCCGTGCGCCGGTCGCCAGCAAAGAAAG
CAAGCTTTCTTCCTGATGCCCCTCGACTTGCATGTGTTAAGCCTGTAGCTAGCGTTCATCCTGAGCCAGGATCAAACTCTTCAT
And a BLAST search indicates that it matches 100% over its entire lenght to the ‘Bacteroides fragilis strain NCTC 9343 16S ribosomal RNA gene, complete sequence’.
As you can see, things may occasionally work even when you are dealing with bioinformatics.
Working with anvi’o standard files
The beauty is that you don’t really need to do anything if you already have been using anvi’o to study your metagenome. All you need to do is to re-run the program anvi-run-hmms on your contigs database, and you are golden. When you visualize your metagenomes with the program anvi-intearctive, you will automatically see where all the rRNA genes are among your contigs. Or you can access to them via the program anvi-get-sequences-for-hmm-hits as demonstrated above.
Here I will download the Infant Gut data pack from this tutorial to demonstrate how things run for an already existing anvi’o project. Let’s go.
# download the data, unpack, and get into the directory:
wget https://ndownloader.figshare.com/files/8252861 -O INFANT-GUT-TUTORIAL.tar.gz
tar -zxvf INFANT-GUT-TUTORIAL.tar.gz
cd INFANT-GUT-TUTORIAL
# run HMMs for the new profile
anvi-run-hmms -c CONTIGS.db
Alright!
Now we run the interactive interface,
anvi-interactive -p PROFILE.db -c CONTIGS.db
You will see the addition of a new layer on the display:
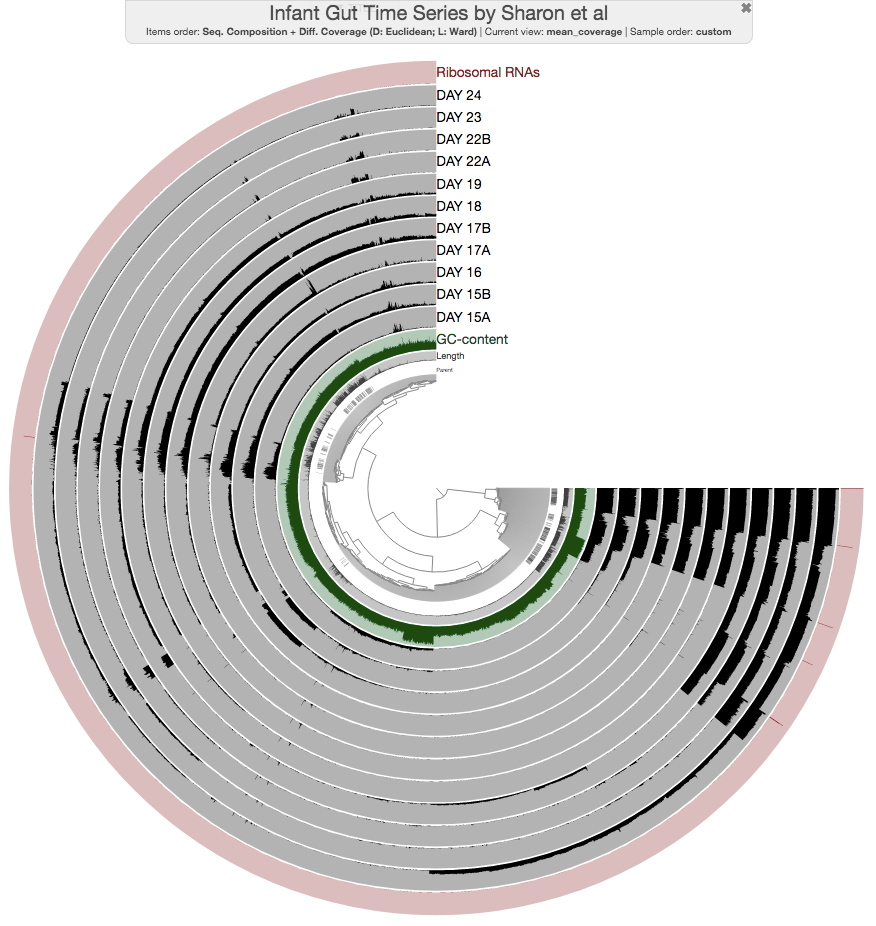
The marks in most outer layer identify contigs with rRNA genes. If you zoom in to one of those, and click ‘Inspect’ from your right-click menu, you can see them clearly marked in red:
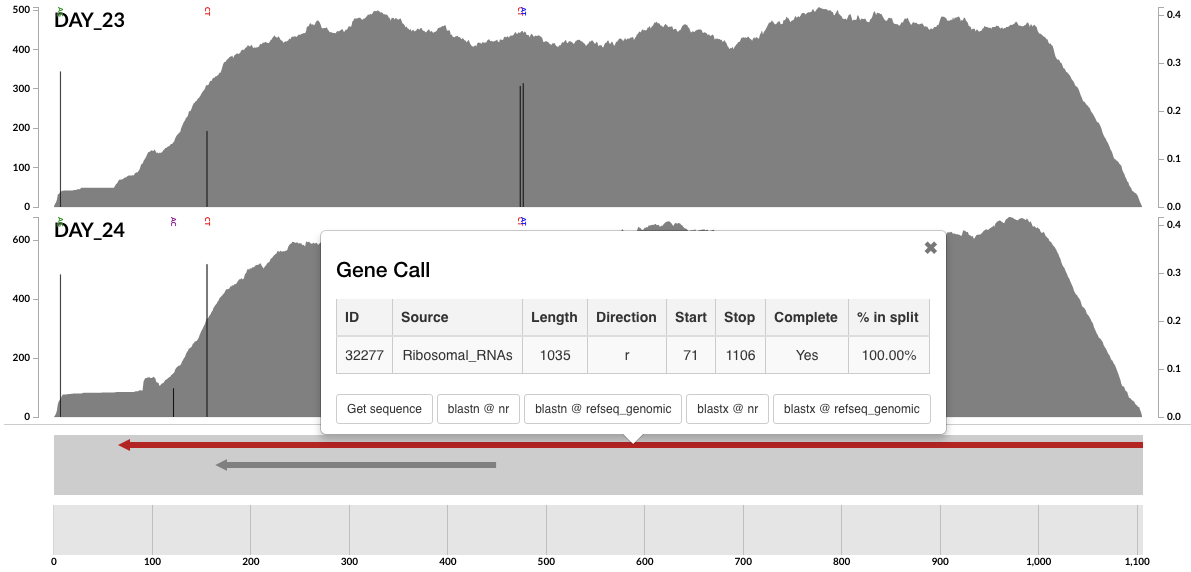
You can get the sequence for it, or go to NCBI with it directly from the interface!
We hope this is useful to you, and please feel free to send any questions to our way as usual.
Under the hood
Here are some technical stuff for archival purposes. You clearly don’t need to read this if you are not interested.
Torsten HMMs to anvi’o HMMs
For posterity, this file how we reformatted HMMs from the Barrnap repository.
Design issues
We had designed the anvi’o framework for HMMs to work with gene calls. For instance, when a new contigs database is generated, first Prodigal identifies all the gene calls (unless the user has used --external-gene-calls directive to provide their own gene calls), and then AA sequences for gene calls are handed over to another anvi’o module anvi-run-hmms uses to search for HMM profiles, say, to identify single-copy core genes. When we wanted to work with rRNA HMMs, we run into three major problems;
-
Torsten’s HMM profiles for rRNAs were obviously in RNA alphabet, so we needed to hand over sequences in RNA alphabet to the anvi’o module that was used to working with AA sequences,
-
Because Prodigal couldn’t identify those genes properly, we no longer could rely on gene calls to run these HMMs, so instead of genes, we now needed to send contigs to this module,
-
Since we no longer are working with gene calls, we all of a sudden had a problem of ‘follow-up’: when you hand over a bunch of gene calls with unique IDs to another module, it is much more easier to make sense of the results when they are back, and link them to the information that is already in the database. But working with contigs was going to take that away.
We solved these things in multiple ways without braking other things in the repository.
First, we added a new property to the standard anvi’o HMM profile files: target.txt. For Campbell et al., which is the good old bacterial single-copy core gene HMMs, the content of target.txt is AA:GENE, telling anvi’o that it wants amino acid sequences for genes. For the new rRNA profile, the same file says RNA:CONTIG. As you can gess, this one tells anvi’o that it is supposed ot be working with contig sequences in RNA alphabet.
We also extended our design to do create new gene calls in the contigs db based on results that are returned from an HMM profile that uses contig sequences. These additions are fully extensible, so if you come up with your own collection of, say, antibiotic-resistance genes, they should work seamlessly. All you need to do is to mimic one of the already-working HMM profiles in the database.
Then we decided to display rRNA gene calls in ‘red’, instad of ‘green’ or ‘gray’, in the inspection pages of the interactive interface. These take some serious coding, but anvi’o is very flexible, and adding features more fun than a nightmare.