



Table of Contents
The latest version of anvi’o is v7.1. See the release notes.
Contents of this article requires anvi’o v5 or later. You can learn which version you have on your computer by typing anvi-profile --version in your terminal.
Guy Leonard has made a script to download any of the BUSCO HMM collections and automatically convert them into Anvi’o ready versions. The script and an example of usage can be found here.
This post describes a collection of 83 single-copy core genes I curated from BUSCO, so anvi’o can identify eukaryotic genome bins in metagenomic binning efforts (stay tuned, viruses, you are not forgotten, and your time will come soon), and estimate their level of completion.
A brief background:
While anvi’o comes with reference single-copy core gene collections to assess completion and redundancy of bacterial and archaeal bins, until v5, it did not include any collection for viruses and eukaryotes. This is a limiting factor for anvi’o users who are interested in the genome-resolved metagenomics workflow to study domains of life beyond Bacteria and Archaea.
Fortunately, it is easy to incorporate new HMM models into anvi’o to search for particular gene families of interest. Here I simply used this opportunity to curate a collection of eukaryotic single-copy core genes for anvi’o using the BUSCO database. This led to a promising (yet preliminary) collection of 83 single-copy core genes that installs with anvi’o since version v5.
BUSCO: Benchmarking Universal Single-Copy Orthologs
Single-copy core gene collections dedicated to different lineages of organisms have been created under the label BUSCO for Benchmarking Universal Single-Copy Orthologs (see the web page). Here are the related articles:
-
BUSCO applications from quality assessments to gene prediction and phylogenomics. doi:10.1093/molbev/msx319
-
BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. doi:10.1093/bioinformatics/btv351
The following sections describe the incorporation of a BUSCO collection of single-copy core genes dedicated to Protists into anvi’o and its subsequent testing using reference genomes and eukaryotic MAGs recovered from three distinct genome-resolved metagenomic projects.
Protists include all eukaryotic cells not affiliated with plants, animals or fungi.
The BUSCO collection of single-copy core genes for Protists
The BUSCO collection of single-copy core genes for Protists (named protists_ensembl) is available from here. It contains HMM models for 215 single-copy core genes.
To test the effectiveness of this collection when used from within anvi’o v4, I generated contigs databases for four complete eukaryotic genomes: Bathycoccus prasinos, Micromonas pusilla, Ostreococcus tauri and Pseudo-nitzschia multistriata.
I followed the relevant tutorial to create an anvi’o collection, and used anvi-run-hmms program to test it on the four reference eukaryotic genomes, using different e-value cut-offs.
BUSCO uses optimized length and e-value cut-offs for each HMM model. This flexibility is not available in anvi’o v4, for which a single e-value cut-off must be defined for all HMM models within a collection.
I detected some of the single-copy core genes dozens of times in all four reference eukaryotic genomes, even after varying the e-value cut-off from E-15 to E-100. Most others were detected once, as expected. To avoid increased number of false positives, I selected a fixed e-value of E-25, and removed genes that occurred multiple times or never in each of the four reference genomes, resulting in a total of 83 single-copy core genes. I combined them in a new collection of single-copy core genes dedicated to protists named BUSCO_83_Protista.
As expected, here are the anvi’o completion estimates for my reference genomes:
| Genome | Completion | Redundancy |
|---|---|---|
| Bathycoccus_prasinos | 100.00% | 0.00% |
| Micromonas_pusilla | 100.00% | 0.00% |
| Ostreococcus_tauri | 100.00% | 0.00% |
| Pseudo-nitzschia_multistriata | 100.00% | 0.00% |
In the following section, I tested the efficacy of this collection to estimate the completion and redundancy of eukaryotic MAGs characterized from different genome-resolved metagenomic projects.
MAG stands for metagenome-assembled genome.
Assessing the completion/redundancy of eukaryotic MAGs from 3 different projects
TARA Oceans
I recently spent some time manually binning ~2.6 million scaffolds assembled from metagenomes of the TARA Oceans project that cover the surface of four oceans and two seas. Among the ~1,000 MAGs we characterized in this project, 45 belong to the domain Eukarya. Until now, their completion had not been tested beyond the use of inappropriate bacterial single-copy core genes.
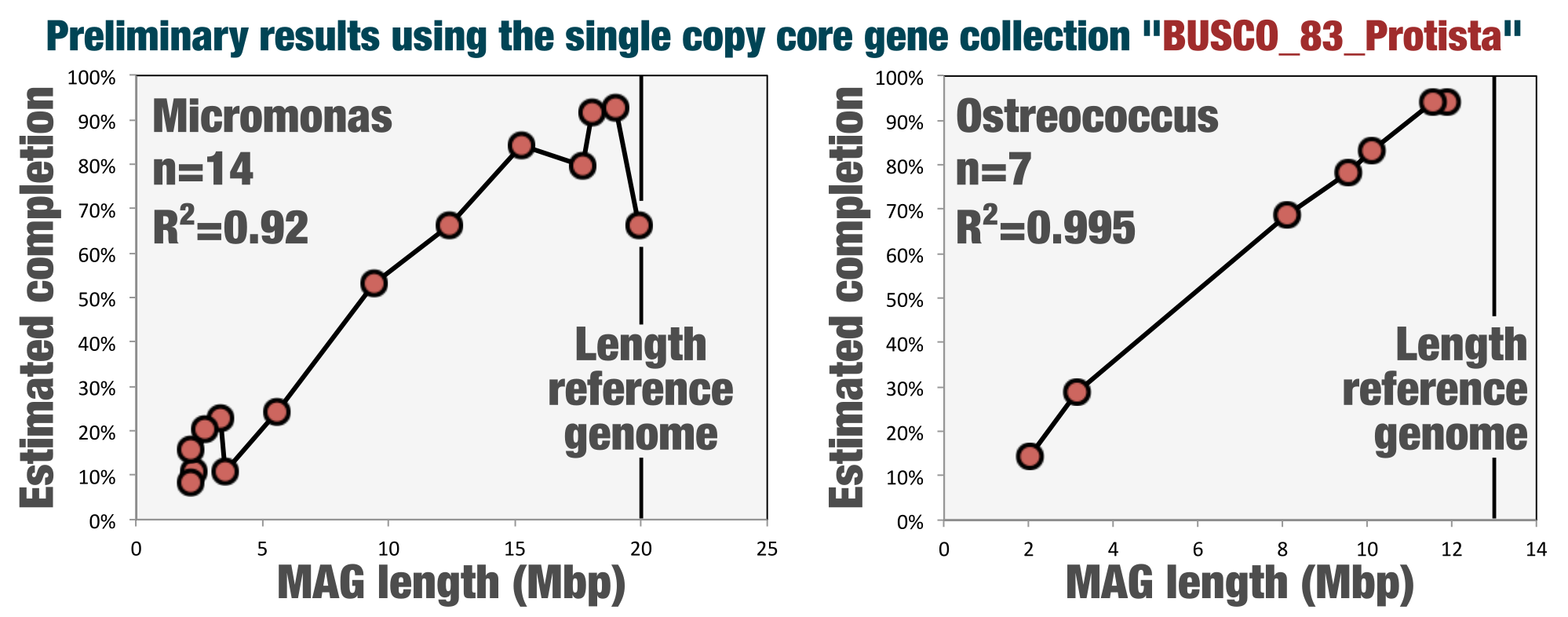
Micromonas
14 TARA Oceans MAGs were affiliated to Micromonas, and we know from cultivation that Micromonas genomes are typically 20 Mbp long. This is a good case study to determine the accuracy of the new collection of single-copy core genes.
Here are the related statistics:
| Eukaryotic_MAG | Length | Genus | Completion | Redundancy |
|---|---|---|---|---|
| TARA_ANW_MAG_00088 | 19,914,044 | Micromonas | 66.27% | 4.82% |
| TARA_ANE_MAG_00101 | 19,009,552 | Micromonas | 92.77% | 4.82% |
| TARA_ASW_MAG_00046 | 18,057,117 | Micromonas | 91.57% | 6.02% |
| TARA_ANW_MAG_00080 | 17,700,320 | Micromonas | 79.52% | 1.20% |
| TARA_RED_MAG_00119 | 15,250,294 | Micromonas | 84.34% | 3.61% |
| TARA_ASE_MAG_00032 | 12,391,309 | Micromonas | 66.27% | 4.82% |
| TARA_ANW_MAG_00074 | 9,401,388 | Micromonas | 53.01% | 1.20% |
| TARA_ANE_MAG_00090 | 5,559,939 | Micromonas | 24.10% | 1.20% |
| TARA_ASW_MAG_00039 | 3,501,361 | Micromonas | 10.84% | 0.00% |
| TARA_ASW_MAG_00034 | 3,334,468 | Micromonas | 22.89% | 1.20% |
| TARA_ANW_MAG_00075 | 2,699,399 | Micromonas | 20.48% | 1.20% |
| TARA_ANW_MAG_00081 | 2,279,971 | Micromonas | 10.84% | 0.00% |
| TARA_ASW_MAG_00037 | 2,179,362 | Micromonas | 15.66% | 0.00% |
| TARA_ANE_MAG_00099 | 2,133,633 | Micromonas | 8.43% | 0.00% |
There is a strong correlation between length and completion estimates (R-scare > 0.9), and MAGs with a length close to 20 Mbp have high completion values (with the exception of TARA_ANW_MAG_00088 for which completion seems a little off - but this can be due to assembly or binning problems).
Ostreococcus
7 TARA Oceans MAGs were affiliated to Ostreococcus (reference genomes are ~13 Mbp), and here are the related statistics:
| Eukaryotic_MAG | Length | Genus | Completion | Redundancy |
|---|---|---|---|---|
| TARA_PSW_MAG_00136 | 11,885,609 | Ostreococcus | 93.98% | 1.20% |
| TARA_ASE_MAG_00036 | 11,537,750 | Ostreococcus | 93.98% | 2.41% |
| TARA_RED_MAG_00118 | 10,110,290 | Ostreococcus | 83.13% | 1.20% |
| TARA_ANE_MAG_00093 | 9,555,746 | Ostreococcus | 78.31% | 2.41% |
| TARA_PON_MAG_00082 | 8,114,168 | Ostreococcus | 68.67% | 2.41% |
| TARA_PSE_MAG_00129 | 3,144,407 | Ostreococcus | 28.92% | 0.00% |
| TARA_RED_MAG_00116 | 2,042,466 | Ostreococcus | 14.46% | 0.00% |
The correlation between the genomic length and completion estimates for Micromonas and Ostreococcus MAGs characterized from the surface ocean was quite promising:
Overall, average redundancy estimates for the 45 eukaryotic MAGs went from ~20% with the bacterial single-copy core gene collection to 1.4% with BUSCO_83_Protista. This suggests the eukaryotic MAGs represent individual population genomes with no substantial amounts of contamination. As a result, it seems key information computed by anvi’o (especially differential coverage and sequence composition) is also effective for the characterization of (sometimes near-complete) eukaryotic genomes. Good.
Pseudo-nitzschia from the Southern Ocean
In another project led by Anton Post, we have characterized using genome-resolved metagenomics a 34 Mbp MAG corresponding to Pseudo-nitzschia. This lineage was prevailing in the Ross Sea polynya in the coast of Antarctica during the 2013-2014 austral summer. The story is not yet published, and until now I had never estimated the completion or redundancy of this MAG.
BUSCO_83_Protista estimated this MAG to be 86.8% complete and 2.4% redundant. This made my day…
Candida albicans in a gut metagenome
Sharon et al. (2013) characterized a MAG corresponding to Candida albicans (a fungi) from metagenomes of an infant gut, and we later recapitulated this finding using our own metagenomic binning effort of the same dataset. The Candida albicans MAG we recovered is 13.8 Mbp long. Based on BUSCO_83_Protista, this MAG is 83.1% complete and 7.2% redundant. A reference Candida albicans genome is 14.7 Mbp long, suggesting that the estimate is not far from what one would expect.
Not too bad given that fungi are not protists. This suggests such collection could work to a certain extent beyond the realm of protists.
Final notes
With this addition, anvi’o can now estimate the completion and redundancy of microbial genomes from three domains of life.
However, please keep in mind that single-copy core gene collections only provide a rough estimation of the completeness and contamination of MAGs. Visualizing a MAG in the context of recruited reads from metagenomes for instance (the environmental signal) remains essential when it comes to genome-resolved metagenomics, and this approach very often allows the detection and removal of suspicious contigs that contain no single-copy core genes and hence are invisible from the perceptive of completion / redundancy estimates. Thus, these estimates are only one of multiple parameters one can (and maybe should) use to characterize, manually refine, and assess the biological relevance of eukaryotic MAGs.
Of course, these problems are less relevant if one is working with cultivar genomes and single cell genomes .. Or are they? (smiley)
Estimating the completion of eukaryotic genomes reconstructed from metagenomes hasn't been quite straightforward.
— A. Murat Eren (Meren) (@merenbey) May 10, 2018
But here is a blog post from @tomodelmont, extending #anvio's capabilities towards that direction 🎉https://t.co/lK43avyi05