Table of Contents
This article describes how to import functional annotations of genes into anvi’o.
Anvi’o also provides a simpler way to annotate your genes using NCBI COGs. If you would like to explore that option, please read this article.
Have you been struggling with importing your functions into anvi’o? You want to use XXX to annotate your functions because everyone says it is awesome, but the output from XXX is not compatible with anvi’o? Let us know, and we will see what we can do!
Anvi’o accepts functional annotation at the gene-level. Gene IDs in your input files should correspond to gene IDs in the contigs database, hence, functional annotation should be done after exporting genes from the contigs database. The basic workflow goes like this: (1) generate your contigs database, (2) export your gene sequences, (3) assign functions to them, and (4) import results back into your contigs database.
Important note: There are many ways to have your genes annotated with functions, but, there is only one way to make sure the gene IDs in the files you generate using external tools correspond to the gene IDs in the database: export your DNA or AA sequences from the anvi’o contigs database you wish to annotate using anvi’o program anvi-get-sequences-for-gene-calls.
Introduction
You will be using anvi-import-functions to do things described here, but before we start with the input file formats here is a little information about the program. You can import functions into anvi’o incrementally. For instance, let’s assume you have annotated your genes with PFAMs, imported those results into anvi’o. Then you decided to do it with TIGRFAMs, too. In fact you can import TIGRFAMs as an additional data into the database without erasing PFAMs. Functions can be imported multiple times from multiple sources without overwriting previous imports. This is the default behavior of the program, but alternatively, you can use the --drop-previous-annotations flag to make a fresh start, in which case all previous imports would be erased from the db, and only the final input will be stored in it.
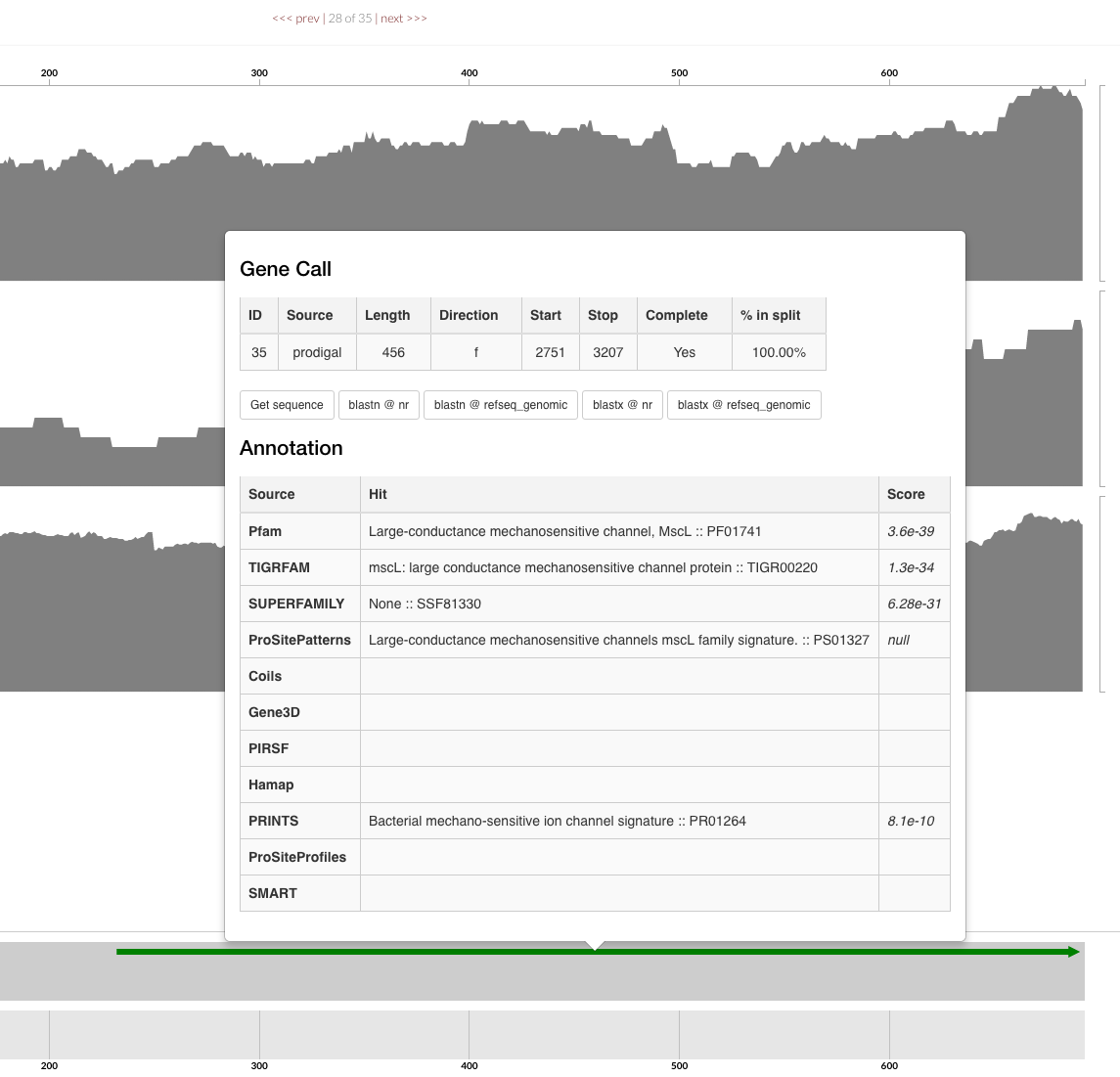
When you import your gene calls you get more comprehensive outputs during summary. Also when you inspect your contigs, you get to click on gene calls and see how they are annotated:
Simple matrix
This is the simplest way to get the functional annotation of genes into anvi’o is to do it through a flat, TAB-delmited text file we call functions-txt. Please see the details of this format using the link.
EggNOG database + emapper
Anvi’o has a parser for emapper output for EggNOG database. After exporting your amino acid sequences from your contigs database,
$ anvi-get-sequences-for-gene-calls -c CONTIGS.db \
--get-aa-sequences \
-o amino-acid-sequences.fa
You can either do it online, through the interface on EggNOG, or locally by following the relevant tutorial on emapper.
Once you have your output file, say amino-acid-sequences.fa.emapper.annotations, you can import it back into the contigs database from which you exported the amino acid sequences:
anvi-script-run-eggnog-mapper -c CONTIGS.db \
--annotation amino-acid-sequences.fa.emapper.annotations \
--use-version 1.0.3
The script is called ‘run’ eggnog mapper, but what it does is to import its results. Well, historically, this script could run emapper on a given contigs database automatically and import results back into it seamlessly without leaving any room for user errors. However, due to the SIGINT and SIGTERM calls in the emapper server code, the software is not quite software friendly and can’t be called from within other software in a straightforward manner. Hence the --annotation flag as a workaround, so you can import your own results, and the weird name for the anvi’o script. Becasue bioinformatics.
The version in the command line above is the version number of emapper you used for annotation (because the number of fields change from one version to the other, and anvi’o needs to know exactly which version you used).
InterProScan
Anvi’o has a parser for InterProScan. To use InterProScan you should first export AA sequences for all your gene calls:
$ anvi-get-sequences-for-gene-calls -c CONTIGS.db \
--get-aa-sequences \
-o amino-acid-sequences.fa
After running InterProScan on this file like this (assuming you have it downloaded):
$ ./interproscan.sh -i amino-acid-sequences.fa \
-f tsv \
-o interpro-output.tsv
You can import results into the database:
$ anvi-import-functions -c contigs.db \
-i interpro-output.tsv \
-p interproscan
That’s it!
Xabier Vázquez-Campos to the rescue
It doesn’t work?
If the InterProScan parser within anvi-import-functions is not working for you, chances are that you used the --iprlookup, --goterms and/or the --pathways options when you ran InterProScan. This happens because the parser expects a table with 11 columns, and you have 15. Columns 12 and 13 are the InterPro integrated annotations, while 14 and 15 correspond to the GOterms and pathway annotations respectively.
Fortunately, there are ways around it. The easiest way it is to remove these extra columns and keep going:
cut -f 1-11 interpro-output.tsv > interpro-output_fix.tsv
anvi-import-functions -c contigs.db \
-i interpro-output_fix.tsv \
-p interproscan
Although this doesn’t handle your precious GOterm annotations or the InterPro cross-references (IPR numbers).
Alternatively, you can use the standard import. Although, you’ll likely have redundant annotations (some protein domains can appear more than once in a single protein).
echo -e "gene_callers_id\tsource\taccession\tfunction\te_value" > table_to_import.tsv
cut -f 1,4-6,9 interpro-output.tsv >> table_to_import.tsv
anvi-import-functions -c contigs.db \
-i table_to_import.tsv
And, unfortunately, this may not work in some cases. E.g. if you got TMHMM or Phobius annotations, the e-value column will be always a dash -, not a number and the standard anvi-import-functions doesn’t like it.
The final solution
To avoid any and all of these problems I created iprs2anvio.sh, a little BASH script that handles:
- InterPro cross-referenced annotations (the 12th and 13th columns) (
-ror--iproption). - Dereplicates multiple matches of the same motif profile to a single protein, keeping the lowest e-value.
- Also handles score-based annotations: ProSiteProfiles and HAMAP, and keeps the annotation with the higher score.
- Handles the dashes
-in the e-value column. - Allows to specify the annotation databases of interest and even split the output, i.e. a single file per database (
-sor--split). - Can extract optional annotations from columns 14 and 15:
- GOterms (
-gor--go_terms). - Pathways (
-por--pathways).
- GOterms (
iprs2anvio.sh -i interpro-output.tsv -o interpro-output -g -p -r
anvi-import-functions -c contigs.db \
-i interpro-output_iprs2anvio.tsv
It is not the most efficient script in the world, but does the job.
Note that iprs2anvio.sh has been developed on Linux and it hasn’t been tested in Mac yet. It requires GNU getopt to handle options, likely in your system already if you use Linux. For Mac, you can install it easily using MacPorts with sudo port install getopt
Prokka
You can use annotations produced by Prokka with anvi’o, however, in order to do that you will need to create a new contigs database, since Prokka has to run on your contigs, and does not have a workaround to work only with gene calls.
A recipe to work with Prokka by Antti Karkman is available here.
KEGG Modules
A tutorial on how to import KEGG modules into an anvi’o contigs database by Elaina Graham is available here.
A better way?
You have better / faster / more accurate ways to do it? Let us know!