
Table of Contents
- On a journey characterizing a thousand microbial genomes from the surface of four oceans and two seas
- The TARA Oceans project provided a challenging dataset for metagenomic binning
- We reconstruct 30.9 billion reads one geographic region at a time
- We characterized and curated more than one thousand MAGs
- Many MAGs were widespread at the surface of the oceans and seas
- Our first in depth analysis focused on nitrogen fixation
- It might take years to fully appreciate the genomic information this database entails
I dedicate this post to (1) all persons involved in the TARA Oceans project, arguably the most ambitious environmental surveys of its time, (2) the Meren Lab for all the excitment and hard work exploring new bioinformatics frontiers, and (3) anvi’o, the platform without which none of this would have been possible.
On a journey characterizing a thousand microbial genomes from the surface of four oceans and two seas
It is time to disclose some aspects of a bioinformatics journey that started as a little side project and was completed 20 months later with the characterization of more than a thousand population-level microbial genomes from the surface of oceans and seas without the need for cultivation. The fact that microbial populations in the surface ocean play a critical role in climate but for the most part remain uncultured suggests this database might be relevant to microbial oceanography. Besides, this journey has influenced our metagenomic binning workflow, and we now have better ways to handle “very” large metagenomic assembly outputs.
Here is a word cloud of the blog:

With the topic of this blog post now introduced, you can either continue reading and relive this bioinformatics journey as viewed from my perspective, or watch this cute video of a research vessel expedition across Antarctica. This is a early way out for the procrastinators :)
The TARA Oceans project provided a challenging dataset for metagenomic binning
From time to time, events you have very little control over can deeply influence the direction of your research. This is one of those stories.
It all started in July 2015, when the TARA Oceans consortium released the most comprehensive sequencing effort of metagenomic samples that originated from four oceans and two seas, and characterized an impressive collection of 40 million non-redundant genes by reconstructing individual metagenomic samples. The authors, however, did not connect these genes to any genomes, limiting our understanding of the geographic distribution, phylogeny, functionality and metabolic contributions of a wide range of microbial populations inhabiting the largest biome on Earth.
At the time, the analysis and visualization platform anvi’o was in the process of being published, and Meren and I were eager to test our new manual metagenomic binning workflow on large and challenging datasets. I immediately started playing with the TARA Oceans data, focusing on a size fraction we anticipated could be prone to good assemblies: the filters enriched in archaeal, bacterial and pico-eukaryotic cells.
We reconstruct 30.9 billion reads one geographic region at a time
We downloaded 93 TARA Oceans metagenomes, which after quality control provided a total of 30.9 billion reads of 100 nucleotides. They cover the surface of the Mediterranean Sea, Red Sea, Atlantic Ocean, Indian Ocean, Pacific Ocean and Southern Ocean. Note that we released a document describing our entire bioinformatics workflow, starting from the download of these metagenomes.
How can one best take advantage of nearly 31 billion reads from the surface ocean? A single metagenomic co-assembly using the entire dataset is a very memory consuming option, even on a large server. In addition, it would lead to a highly complex puzzle to solve, with the mixing of closely related genomes that might not overlap in their niche partitioning. Thus, we decided to perform 12 independent co-assemblies based on the geographic coordinates of metagenomes. The rationale of doing this was that metagenomes originating from the same geographic region (e.g., the Mediterranean Sea) were more likely to overlap in the sequence space, increasing the mean coverage (and hence the extent of reconstruction) of microbial genomes during the co-assembly.
Here are the 12 geographic regions we used:
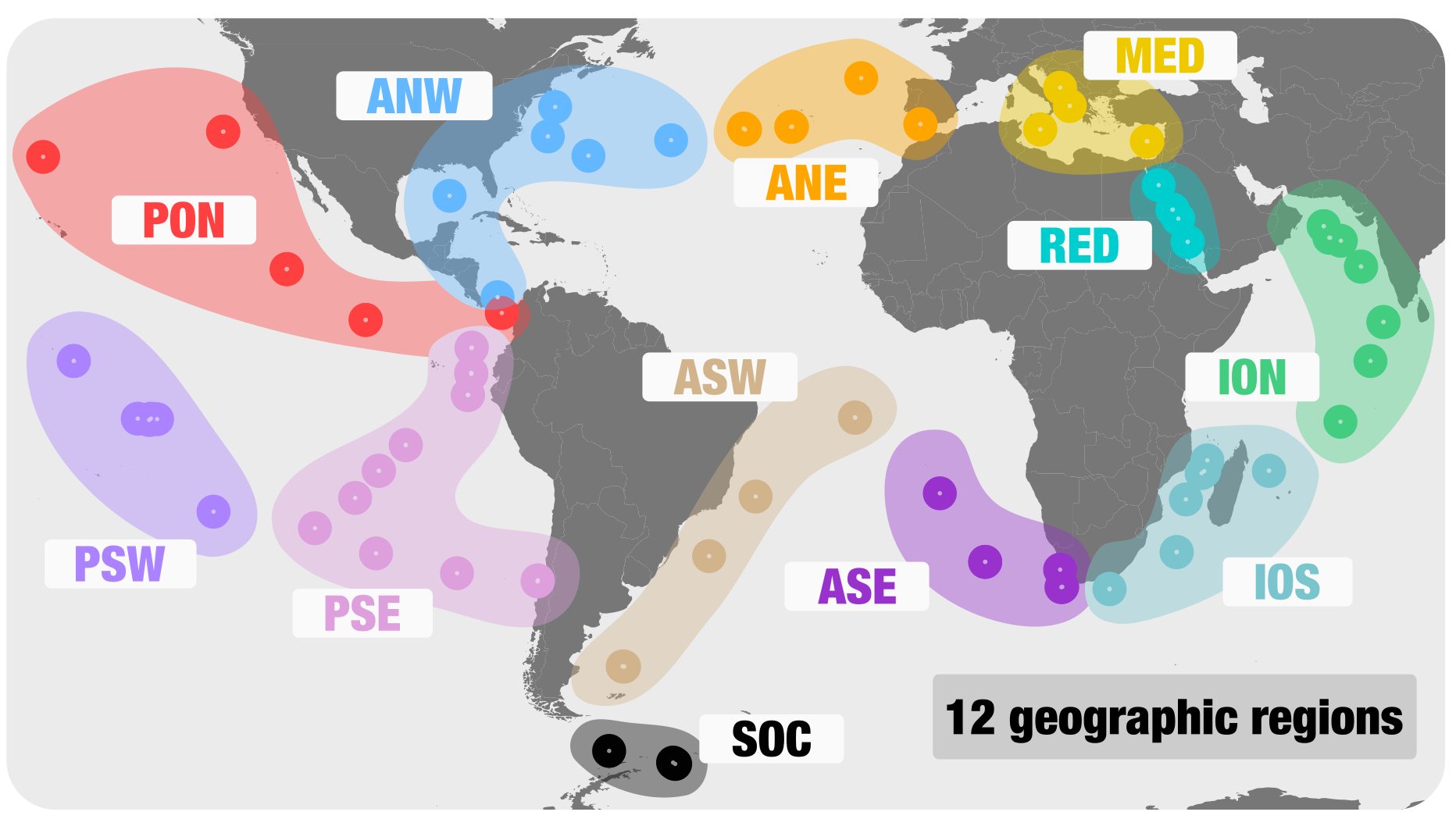
We generated 2.6 million scaffolds >2.5 kbp from the 12 geographically bound co-assemblies. Note that the assemblies were performed on a large server Christopher Quince granted us access to. One easy way to deal with these scaffolds would be to run an automatic binning algorithm in order to generate bins. Automatic binning is trendy and has been used to support the analysis of many metagenomic surveys in the past few years. However, while these algorithms are often described as highly accurate, we found that they all make evident mistakes, even when running on very simple metagenomic datasets. You can visit this anvi’o tutorial to learn more about this problem. As a result, we had no choice but to take the hard road of manually binning these 12 large metagenomic assembly outputs…
In the following months, our failures and few successes at processing these scaffolds have pushed the boundaries of anvi’o and shaped our manual binning workflow in many ways. In particular, we have dramatically improved the efficiency of scaffolds profiling to cope with large assembly outputs and minimized steps during manual binning for an optimal user experience. In addition, we also developed a strategy to combine binning results from multiple co-assemblies while removing genomic redundancies (thanks, Alon!). Finally, Meren found enough time and energy to create a long awaited program (lacking inspiration, he called it anvi-split) to generate self-contained anvi’o files focused on individual bins for targeted analyses, visualizations and sharing of genomic data.
Eventually, I manually characterized 30,244 bins with minimal redundancy in single-copy genes. This describes pretty well how I felt when starting the manual binning for the first geographic region (it was the Mediterranean Sea), and after completion of the binning and curation effort nearly one year later:

We characterized and curated more than one thousand MAGs
While most of the bins were relatively small, a subset appeared to represent large pieces of microbial genomes. Overall, I identified and curated a database of 1,077 metagenome-assembled genomes (MAGs) from the pool of bins. Note that although a labor-intensive step, we consider curation to be critical to the binning workflow, and recommend people not to skip it. For this project (and many others), I used the anvi’o program anvi-refine, which offers an holistic interactive interface to visualize and manipulate scaffolds in the context of various layers of information, as described in this blog, this blog, this blog, ah, and also this blog! Yes, we like blogs very much. This is the first time we have reached the iconic number of 1,000 high-quality MAGs in a single project, and we anticipate this to become more and more common in the years to come.
We analyzed the MAGs and soon realized that some of them where highly similar (with an Average Nucleotide Identity reaching in some cases to 99.99% over most of the genomic length). Of course, this occurred only between MAGs characterized from different co-assemblies, as this level of redundancy collapses within each co-assembly. After the identification of redundant MAGs (requirement: >99% ANI over 50% of the genomic length and Pearson correlation >0.9), we built a final non-redundant database of 957 MAGs (120 MAGs defined as redundant were then only used as genomic replicates). In our first preprint describing this database, Figure 1 displays the number of non-redundant MAGs we characterized for each of the 12 geographic regions:
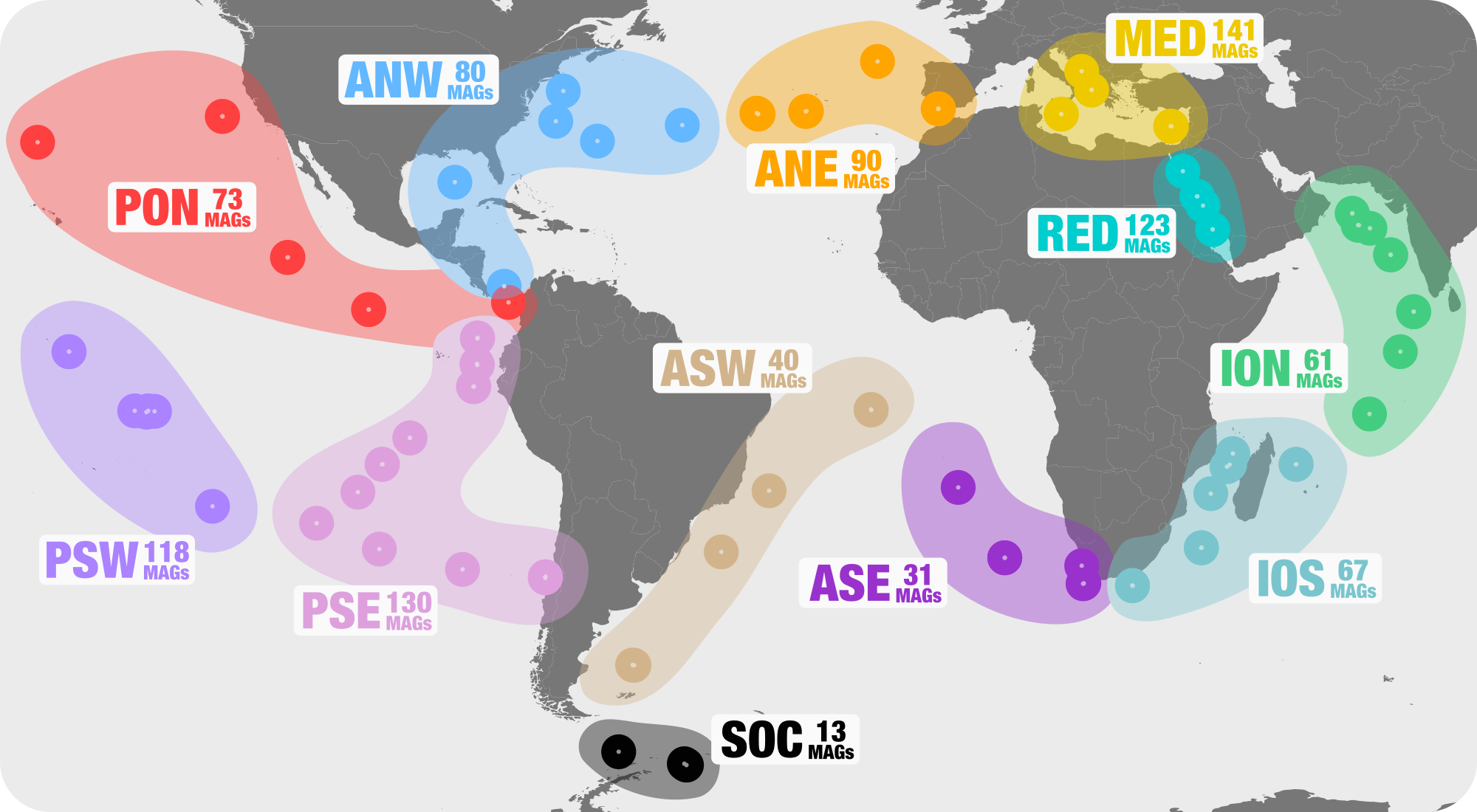
This non-redundant database contained MAGs belonging to the domains Bacteria (n=820), Eukaryota (n=72) and Archaea (n=65), and has been circulating to some extent as a tweet and a bioRxiv preprint. Altogether, these MAGs recruited about 7% of the 30.9 billion reads, suggesting that the genomic content of most marine microbial populations have yet to be characterized. This will not come as a surprise to most marine microbial ecologists.
Many MAGs were widespread at the surface of the oceans and seas
From the recruitment of reads we could determine the relative distribution of each non-redundant MAG in the 93 metagenomes, which provided assess to the niche partitioning of about a thousand microbial populations inhabiting the surface ocean.
To exemplify the results, here’s a display of the geographic distribution for the five most abundant MAGs in the TARA Oceans metagenomes:
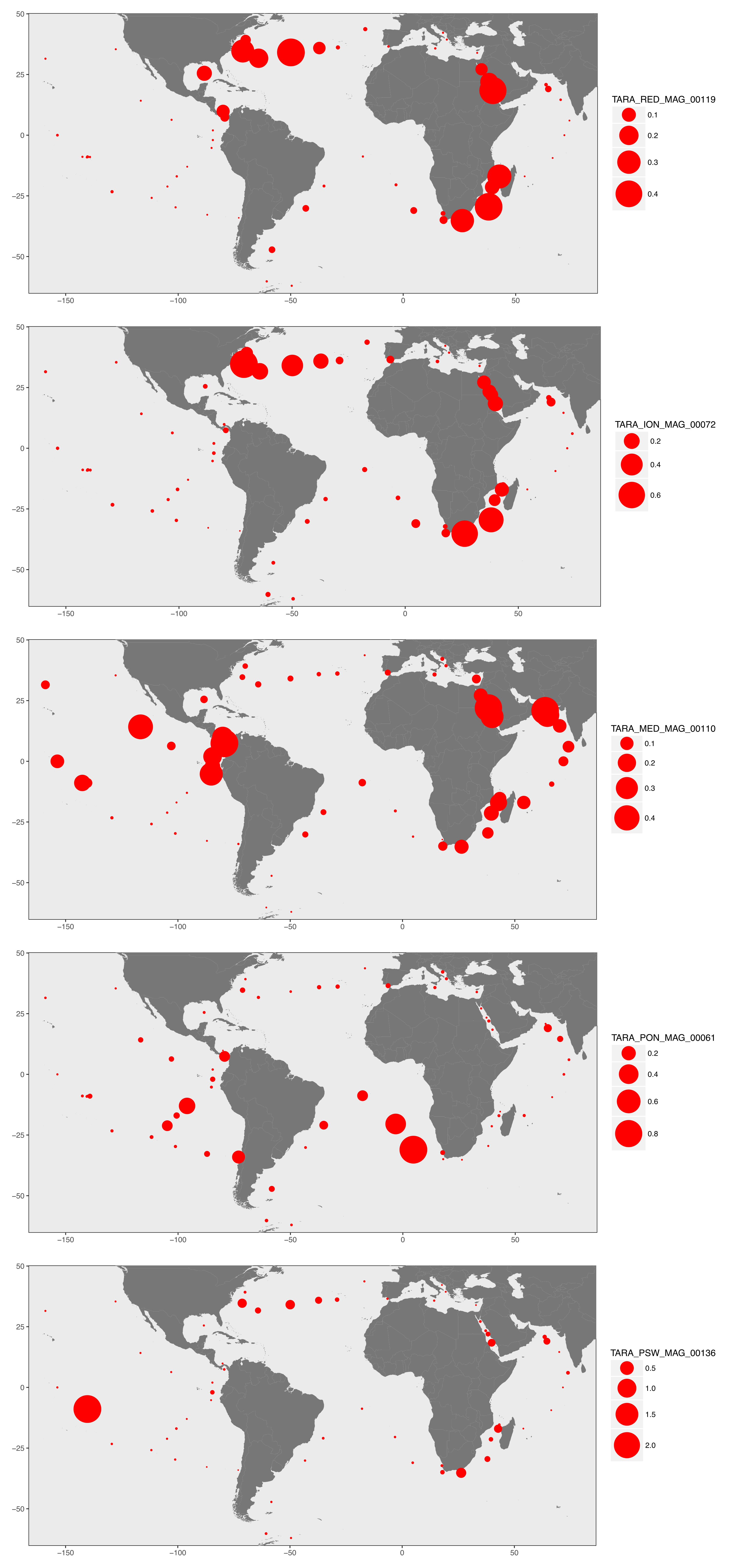
Distribution patterns offered some valuable perspectives, and indicated that many genomes were widespread and occurred in multiple regions (explaining why we identified a non-negligible number of redundant MAGs). These results (1) stress the need to minimize the importance of the coordinates marine microbial cultures are isolated from (especially for the naming of new species), and (2) suggest that in many cases dispersal limitation does not seem to play a major role in the niche partitioning of microbes in the surface of oceans and seas.
Some have suggested that temperature can explain to a large extent the community structure of microbial communities in the surface of the largest biome on Earth. Our results conflict with this notion, as temperature could not explain the distribution patterns of most of the MAGs we have characterized. The dynamic of marine microorganisms is complex, and I like to think that our culture-independent database of hundreds of microbial genomes could contribute to this understanding, maybe by connecting the dots between phylogeny, functioning, micro-diversity and geographic distributions. Anyone interested?
Our first in depth analysis focused on nitrogen fixation
In our first in depth analysis of the database, we focused on 10 non-redundant MAGs containing all the genes required to perform nitrogen fixation. Here is a brief introduction of the study:
Nitrogen fixation impacts the global climate by regulating the microbial primary productivity and the sequestration of carbon through the biological pump. The current dogma is that (1) a few cyanobacterial populations are the main suppliers of the bio-available nitrogen in the surface ocean (multiple culture isolates are available), and (2) non-cyanobacterial populations able to fix nitrogen are rare but diverse (this as only been demonstrated using molecular surveys of the nitrogenase reductase gene, without genomic evidence to substantiate these observations).
We provided genomic evidence for the occurrence in the surface ocean of nitrogen-fixing populations affiliated with Proteobacteria and Planctomycetes. As a cool side note, we provided an interactive interface describing the phylogeny of the Proteobacteria and Planctomycetes MAGs we characterized, with an emphasis on nitrogen-fixing populations. While the phylum Planctomycetes has never been linked to nitrogen fixation prior to our study, this discovery was overshadowed by what the geographic distribution of these genomes have revealed to us: nitrogen fixing populations not affiliated with Cyanobacteria are not only diverse but also abundant in the surface ocean, occasionally across wide ecological niches spanning a large range of temperatures. Here is the Figure 4 in our preprint that describes the abundance of these MAGs:
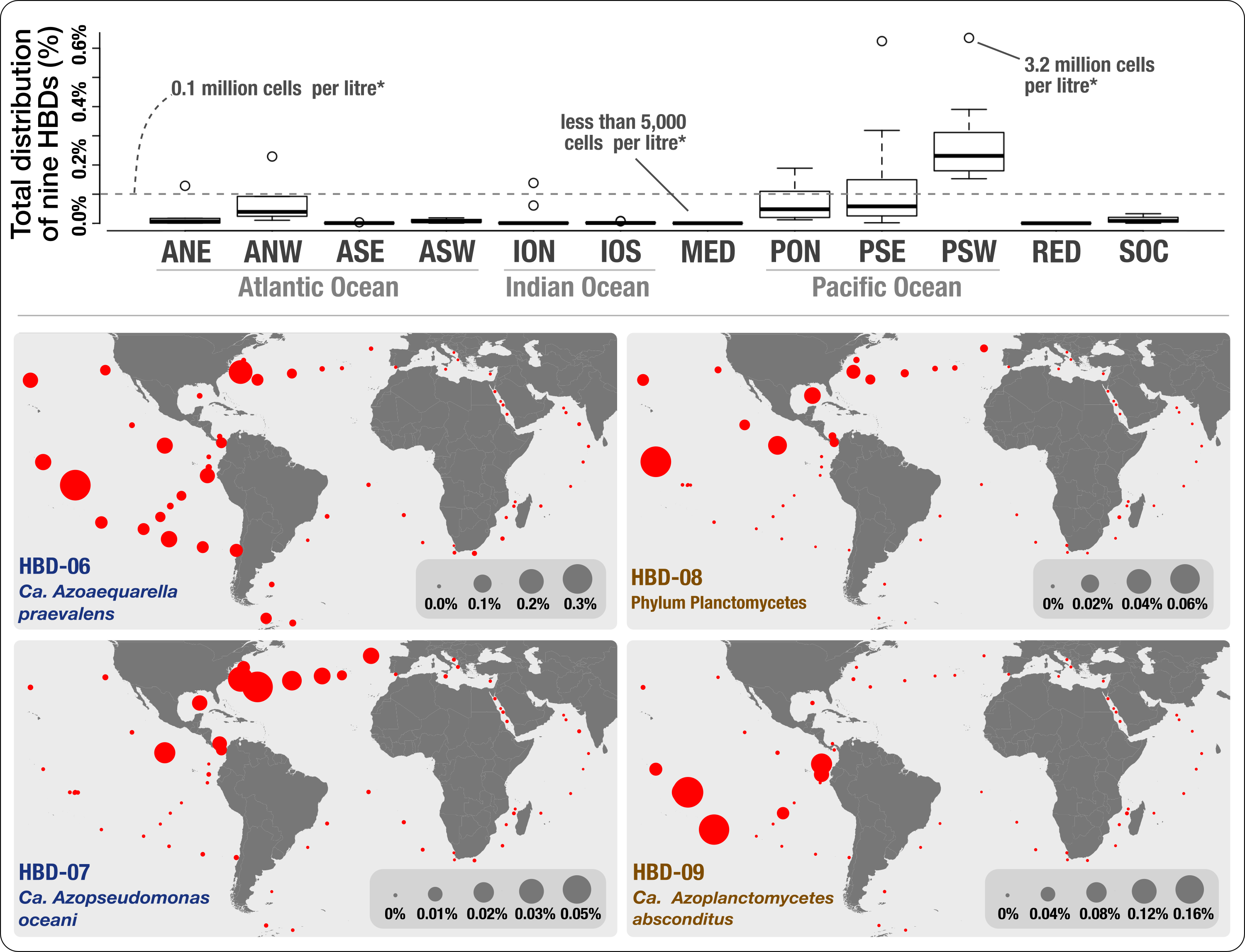
Overall, our analysis conflicted with past molecular surveys that relied on PCR primers prone to methodological biases (keeping in mind that these surveys have been instrumental in our understanding of the diversity of nifH genes in the surface ocean), and emphasized the need to re-assess the role of heterotrophic bacteria in oceanic primary productivity and the nitrogen cycle.
It might take years to fully appreciate the genomic information this database entails
Nitrogen-fixing genomes are not the only MAGs that have catch our eyes in this database. For instance, we are now in the process of describing a new phylum (‘Candidatus Tarascapha’) prevalent in the surface ocean, as well as new Archaeal lineages and SAR11 clades. We also have a few other observations that might be worth exploring in greater details. However, we are trying to motivate researchers to explore various aspects of the database, with or without our help. It might take years to fully appreciate the genomic information this database entails, and we have no interest in doing so alone!
All our analyses are fully available. They include the functional potential, taxonomy and geographic distribution of all the MAGs. One might be tempted to focus on a particular functional family (e.g., Proteorhodopsin), a lineage of particular interest (e.g., Micromonas), or use the entire database to develop exciting bioinformatics tools.
Here is a taxonomic cloud of the database, for the curious:

Now, I have made my little contribution to the TARA Oceans project, and can move on to my other projects. You know, the ones I would have completed months ago if I did not have pursue this little side project…