


Table of Contents
This tutorial is for v6 and later versions of anvi’o. You can identify which version you have on your computer by typing anvi-self-test --version in your terminal.
The latest version of anvi’o is v7.1. See the release notes.
Introduction
Some genetic analyses call for the comparison of specific genetic loci between genomes. For example, one may be interested in investigating evidence for adaptive evolution of the lac operon between different E. coli strains, and the first step to this analysis would be to extract the various lac operon from a collection of E. coli genomes.
To address this example and other genomic loci analyses alike, we present anvi-export-locus, an anvi’o program that enables you to target regions of interest across genomes and/or metagenomic assemblies, and report sequences and/or anvi’o contigs databases for cut loci for downstream analyses.
Briefly, anvi-export-locus cuts out loci using two approaches: default-mode or what we call flank-mode. In the default-mode, the tool locates a designated anchor gene, then cuts upstream and downstream based on user-defined input. Notice that what is “upstream” and what is “downstream” is determined according to the direction of the anchor gene, i.e., if the anchor gene is in the reverse direction, then “upstream” would mean genes that have higher gene callers ids, and vice versa. On the other hand, flank-mode finds designated genes that define the left and right boundaries of the target locus, then cuts in between them. Genes to locate locus anchors or flanking genes are defined through their specific ids in anvi’o or through search-terms that query functional annotations or HMM hits stored in your contigs database.
The purpose of this article is to demonstrate the functionality of anvi-export-locus using a simple and reproducible example: extracting the lac operon from the larger genomic context of E. coli genomes.
Using anvi-export-locus with external gene calls
If you are using external gene calls when generating your contigs databases, then you should make certain genes that are next to each other on a contig, have sequential gene caller ids. If you are using Prodigal, then you have nothing to worry, since this is how Prodigal behaves and this is how we tested anvi-export-locus. To clarify this point, the way anvi-export-locus finds the genes for it to cut is by using the gene callers id. For example, if you are flank-mode and the upstream and downstream genes that you provided hit gene caller ids 40 and 50, respectively, then anvi-export-locus will return the eleven genes with ids between 40 and 50. Intuitively, you would expect these to be next to each other on the chromosome, but there is nothing stopping you from using external gene calls that don’t abide by this intuition, and anvi’o is not going to check whether that is the case or not.

Downloading E. coli genomes
First, let’s download Genbank files for a few representative E. coli strains:
Please see the tutorial here describing how to automagically download genomes from NCBI and include them in anvi’o workflows. We’ll be using this tool to download a few E. coli genomes.
# Set working directory variable for later
WD=$(pwd)
# Download Genbank files
ncbi-genome-download bacteria \
--assembly-level chromosome,complete \
--genus Escherichia \
--metadata metadata.txt \
--refseq-category reference
Next, we’ll make FASTAs, external gene calls, and functional annotations for all the Genbanks we just downloaded:
anvi-script-process-genbank-metadata -m metadata.txt \
--output-dir ecoli \
--output-fasta-txt fasta.txt
Just to have an idea about what is going on, please take a look at the output file ecoli.txt. We will use this file to create our contigs databases for these genomes.
Generate contigs DBs
Now we need to get the fasta files into an anvi’o friendly format. There are many ways to turn your FASTA files into anvi’o contigs databases, but here we will follow our best practices and process all our files using the anvi’o contigs workflow.
First, make a config file for the anvio workflow called config.json:
anvi-run-workflow -w contigs --get-default-config config.json
Then run the contigs workflow!
This step may take a while depending on your computational resources. If you have any questions about running anvi’o workflows please refer to this tutorial here. If you access to an HPC or cluster computer, check out additional parameters here. HINT: you will probably need to edit config.json by switching the rule anvi_gen_contigs_database parameter --ignore-internal-stop-codons to true.
anvi-run-workflow -w contigs \
-c config.json \
--additional-params \
--jobs 6 \
--resources nodes=6
Extract lac operon
We should now have contigs DBs for our genomes.
Now that we have six representative E. coli examples, we will use a combination of the default-mode and flank-mode to cut out the genomic neighborhood around the lac operon and then trim the contig to just contain the target operon.
First, we will use default-mode. This requires the user to provide a --search-term and --num-genes parameter. The --search-term will act as an anchor gene to locate the locus within the contigs provided. In this case, we will use the lacZ gene to locate the lac operon in our E. coli genomes. Once the anchor gene is located --num-genes X,Y will instruct anvi-export-locus to cut X gene(s) upstream and Y gene(s) downstream of the designated anchor gene.
Let’s get cutting!
Default mode
First, we’ll use default-mode to extract the general genomic neighborhood around the lac operon. To cut the lac operon from a single genome, we would have run this command in this general form:
anvi-export-locus -c MY_GENOME.db \
--num-genes 10,10 \
--search-term "lacZ" \
-O MY_GENOME_lac_locus
But since we have multiple genomes we wish to study all at once, we will build a for loop in BASH to make life easier (while making everything more reproducible, and less error-prone at the same time):
mkdir 03_LOCI
cd 03_LOCI
for GENOME in `ls "${WD}"/02_CONTIGS/*-contigs.db`;
do
FNAME=$(basename "${GENOME}" -contigs.db)
anvi-export-locus -c "${GENOME}" \
--num-genes 10,10 \
--search-term "lacZ" \
-O "${FNAME}"_lac_locus;
done
--search-term is NOT case sensitive unless you surround your term in quotes (e.g. --search-term "lacZ")
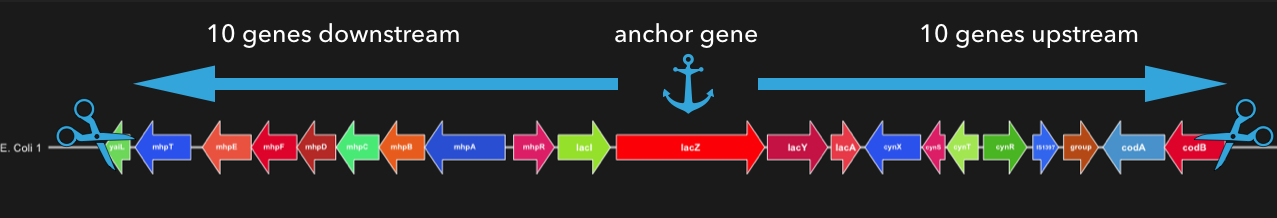
Here is a visual representation of how anvi-export-locus found the anchor gene “lacZ” the cuts 10 genes upstread and downstream.
Flank-mode
Awesome, now we have some smaller contigs that contain the lac operon. BUT, we also grabbed some extra genes that don’t belong to the operon. Let’s use --flank-mode to trim the loci to just contain the lac operon.
To do this, give anvi-export-locus two flanking search-terms: lacI and lacA
for GENOME in `ls "${WD}"/03_LOCI/*.db`;
do
FNAME=$(basename "${GENOME}" _lac_locus_0001.db)
anvi-export-locus -c "${GENOME}" \
--flank-mode \
--search-term "lacI","lacA" \
-O "${FNAME}"_lac_locus_clean;
done
--flank-mode requires flanking genes to be single copies in the contig it’s searching. If your locus of interest does not have fixed coordinates in your genomes or metagenomes, you may need to adjust the -search-terms on a case by case bases.
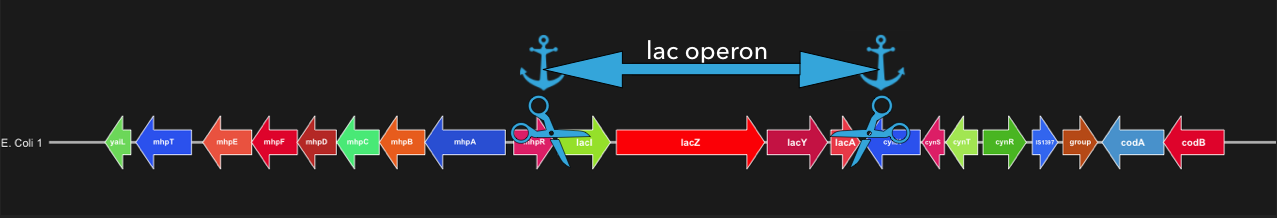
Here is a visual representation of how flank-mode cuts out a locus using flanking genes.
Conclusion
anvi-export-locus is a flexible tool that allows you to extract genomic loci from genomes and metagenomes. In this tutorial, we looked at the classic lac operon in E. coli genomes, but this tool can also be unleashed on any loci in genomes or metagenomic assemblies.
We hope you find amazing applications for this tool. If you have a suggestion or question please do not hesitate to contact us. Also, please report any bugs as an issue on the anvi’o Github repository.