

Table of Contents
This tutorial was written using anvi’o v5.4.
We often need to mix new genomes (whether they are isolate, single-cell, or metagenome-assembled genomes) with those that are already reported and available through the public repositories to generate better comparative insights through phylogenomics or pangenomics. One common public resource for genomes is the NCBI. However, downloading genomes of interest from the NCBI and incorporating the GenBank-formatted public genomes into anvi’o analyses require extra steps.
The purpose of this tutorial is to describe a flexible workflow to download genomes from the NCBI and process them with anvi’o. If you have any questions, please get in touch with us and/or other anvians:

The tutorial will primarily walk you through the steps of downloading genomes of interest from the NCBI (using ncbi-genome-download by Kai Blin), processing NCBI GenBank files (using anvi-script-process-genbank-metadata) to get anvi’o compatible files, and running anvi’o contigs workflow to generate a contigs database for each of these genomes (using anvi-run-workflow).
You can run pip install ncbi-genome-download in your anvi’o environment to install the program that will be essentials for the next steps.
There is another blog post by Mike Lee covering a similar topic for pangenomics. If you are interested in quick and large-scale phylogenomics analyses, you may also want to consider GToTree.
If you have GenBank files from other sources than the NCBI, you may want to take a look at the program anvi-script-process-genbank to generate a FASTA file from it along with an external gene calls file and functions that can be imported into anvi’o.
A simple example
Let’s start with a simple example and assume that you wish to download all complete genomes in the genus Bifidobacterium from the NCBI and turn them into anvi’o contigs databases. An anvi’o contigs database can be used in many ways and in many different workflows since it is quite a central piece of all things anvi’o:
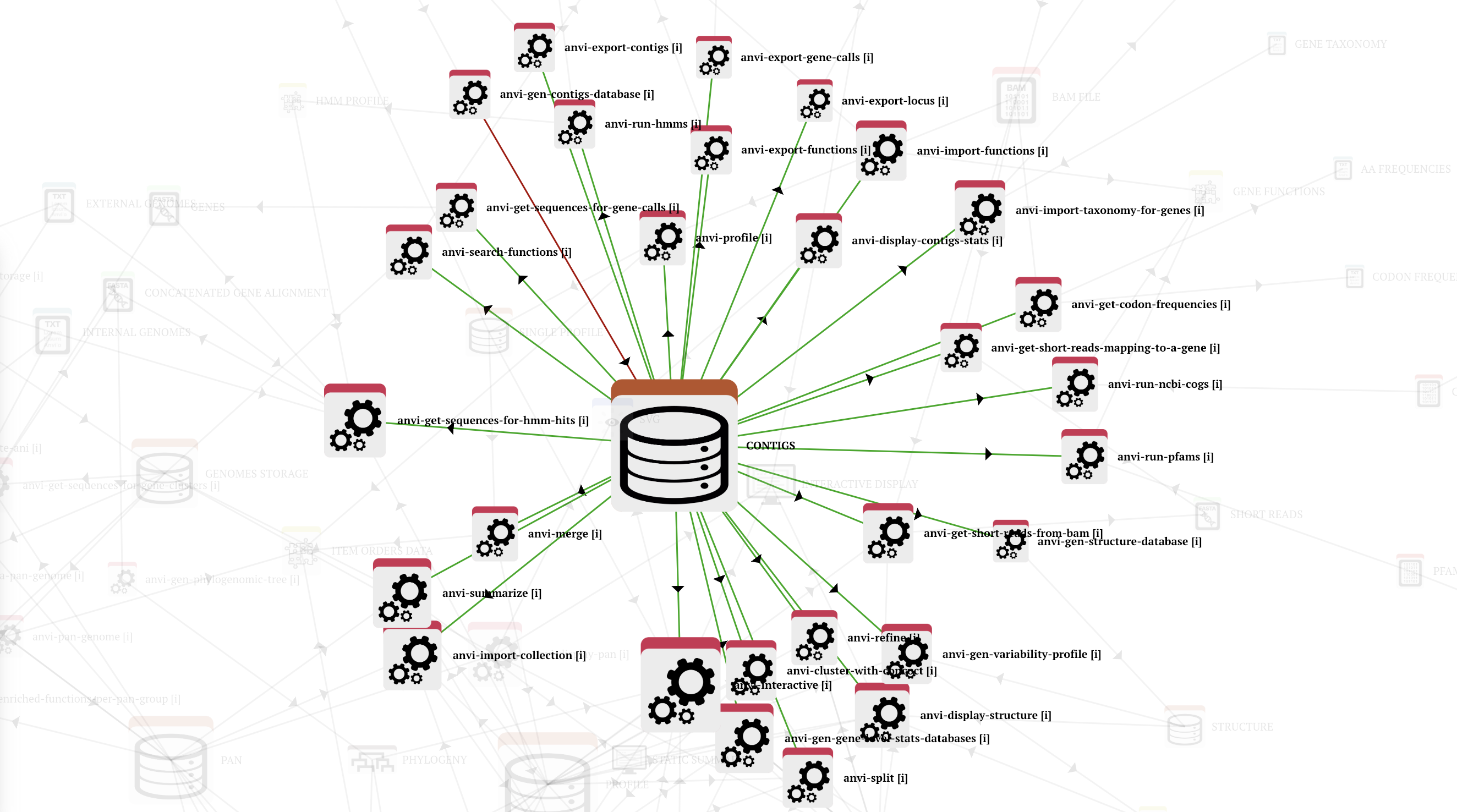
You could download all complete Bifidobacterium genomes like this:
ncbi-genome-download bacteria \
--assembly-level chromosome,complete \
--genus Bifidobacterium \
--metadata NCBI-METADATA.txt
Indeed there are many parameters in the help menu of this program for you to explore.
A successful completion of this command will result in a directory full of GenBank files. Then you can run this command to ask anvi’o to create from each of those files a FASTA file, an external gene calls file, and a functional annotations file:
anvi-script-process-genbank-metadata -m NCBI-METADATA.txt \
--output-dir Bifidobacteria \
--output-fasta-txt fasta.txt
Once you are here, the resulting fasta.txt can go into anvi’o snakemake workflows to generate an anvi’o contigs database for each genome. In its simplest form, you would need to create a config file contigs.json that looks like this:
{
"fasta_txt": "fasta.txt"
}
And then run the anvi’o contigs workflow the following way:
anvi-run-workflow -w contigs \
-c contigs.json
Please consider reading the anvi’o snakemake workflows tutorial to see how you could continue with pylogenomic or pangenomic analyses.
The remainder of this example is here only for educational purposes so you have an idea about the raw steps to accomplish things in anvi’o so you can take better advantage of its Lego-like architecture. This is basically to delay your inevitable evolution into those people in WALL-E due to the luxurious convenience of anvi’o snakemake workflows.
So if you were to be interested in generating your anvi’o contigs databases without using the snakemake workflows, you could run this batch file:
# first create an empty external genomes file:
echo -e "name\tcontigs_db_path" > external-genomes.txt
# loop through fasta.txt (but skip the first line) and assign each of the four
# columns in that file to different variable names for each iteration (the human-readable
# name of the contigs database, the path for the FASTA file, external gene calls file, and
# external functions file:
awk '{if(NR!=1){print $0}}' fasta.txt | while read -r name fasta ext_genes ext_funcs
do
# generate a contigs database from the FASTA file using the
# external gene calls:
anvi-gen-contigs-database -f $fasta \
--external-gene-calls $ext_genes \
--project-name $name \
-o Bifidobacteria/$name.db \
--skip-mindful-splitting
# import the external functions we learned from the GenBank
# into the new contigs database:
anvi-import-functions -i $ext_funcs \
-c Bifidobacteria/$name.db
# run default HMMs to identify single-copy core genes and ribosomal
# RNAs (using 6 threads):
anvi-run-hmms -c Bifidobacteria/$name.db \
--num-threads 6
# add this new contigs database and its path into the
# external genomes file:
echo -e "$name\tBifidobacteria/$name.db" >> external-genomes.txt
done
Once this is done, you have an external genomes file, which is one of the standard inputs for phylogenmic analyses and pangenomic analyses in anvi’o.
A realistic example
The example above covered a simple, but less likely use case. Many of us work with MAGs that often come from understudied branches of life with little taxonomy or complete genomes, and we often want to compare them to other available genomes on NCBI reported by other groups for phylogenomic or pangenomic insights.
Recovering genome IDs to download
In these more realistic use cases, Joe R. J. Healey’s helper script in the ncbi-genome-download distribution, gimme_taxa.py, becomes handy. This helper script allows us to learn specific taxon identifiers for a set of genomes based on hierarchical taxonomic information (i.e., all genomes that belong to a family, or class, and so on).
Say we have a MAG that we were able to assign to the candidate phylum Gracilibacteria (formerly GN02).
To download all genomes affiliated with Gracilibacteria from the NCBI, we will need a specific TaxID, a unique number that translates to a node in the tree of taxa. In this case the node will be Gracilibacteria, and it will allow us to ask gimme_taxa.py to give us all specific IDs for genomes that are under it.
There are two ways to do it. One way to do it is to use the name ‘Gracilibacteria’ as a starting point:
gimme_taxa.py Gracilibacteria \
-o TAX-IDs-RICH.txt
Another way to do the same thing is to first go to NCBI’s Taxonomy Browser and find out the parent TaxID by simply searching for the taxon name of interest. If you do that for Gracilibacteria, you can see that the parent TaxID for this taxon is 363464. We can run the helper script also with that number to find all that descend from there:
gimme_taxa.py 363464 \
-o TAX-IDs-RICH.txt
Please note. These two alternatives will yield identical output files in this particular case. However, using the parent TaxID information explicitly from the NCBI may guarantee that you get exactly what you want. So please pay extra attention to this step and take a careful look at the resulting output file first.
Let’s look into the output:
$ head TAX-IDs-RICH.txt
parent_taxid descendent_taxid descendent_name
363464 363504 uncultured Candidatus Gracilibacteria bacterium
363464 1130342 Gracilibacteria bacterium JGI 0000069-K10
363464 1130343 Gracilibacteria bacterium JGI 0000069-P22
363464 1151650 Gracilibacteria bacterium canine oral taxon 291
363464 1151651 Gracilibacteria bacterium canine oral taxon 323
363464 1151652 Gracilibacteria bacterium canine oral taxon 364
363464 1151653 Gracilibacteria bacterium canine oral taxon 394
363464 1226338 Gracilibacteria bacterium oral taxon 871
363464 1226339 Gracilibacteria bacterium oral taxon 872
While this file is important to survey and keep as a report for later, to make it compatible with what ncbi-genome-download wants, we just need to keep the list of TaxIDs (the descendant taxon ids in this case). For that, you can re-run the helper script this way:
gimme_taxa.py Gracilibacteria \
-o TAX-IDs-SIMPLE.txt \
--just-taxids
Downloading genomes from the NCBI
We can now download all of these genomes:
Pro tip: if you want to first see a list of what is going to be downloaded use the -n flag for a dry run.
ncbi-genome-download -t TAX-IDs-SIMPLE.txt \
bacteria \
-o GN02 \
--metadata NCBI-METADATA.txt \
-s genbank
We specified -s genbank since there are no Gracilibacteria genomes in RefSeq, which is the default database ncbi-genome-download considers, but there are in GenBank. To learn more about the parameters, refer to the help menu of ncbi-genome-download: ncbi-genome-download -h
Processing NCBI genomes
And now for the most beautiful part, once you are done downloading the files, a metadata file is also created by ncbi-genome-download.
Here is a glimpse to this metadata file:
$ column -t NCBI-METADATA.txt | head -n 3
assembly_accession bioproject biosample wgs_master excluded_from_refseq refseq_category relation_to_type_material taxid species_taxid organism_name infraspecific_name isolate version_status assembly_level release_type genome_rep seq_rel_date asm_name submitter gbrs_paired_asm paired_asm_comp ftp_path local_filename
GCA_000404985.1 PRJNA192474 SAMN02441616 ASNE00000000.1 derived from single cell na 1130342 1130342 Gracilibacteria bacterium JGI 0000069-K10 strain=JGI 0000069-K10 latest Contig Major Full 2013/06/03 ASM40498v1 DOE Joint Genome Institute GCF_000404985.1 identical ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/404/985/GCA_000404985.1_ASM40498v1 ./GN02/genbank/bacteria/GCA_000404985.1/GCA_000404985.1_ASM40498v1_genomic.gbff.gz
GCA_000404725.1 PRJNA192390 SAMN02440944 ASND00000000.1 derived from single cell ; partial na 1130343 1130343 Gracilibacteria bacterium JGI 0000069-P22 strain=JGI 0000069-P22 latest Contig Major Partial 2013/06/03 ASM40472v1 DOE Joint Genome Institute na na ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/404/725/GCA_000404725.1_ASM40472v1 ./GN02/genbank/bacteria/GCA_000404725.1/GCA_000404725.1_ASM40472v1_genomic.gbff.gz
Anvi’o includes a program that can process this metadata file and generate a special file, fasta.txt, that is compatible with anvi’o workflows to turn these FASTA files into contigs databases or more. Here is how you would typically run this script:
anvi-script-process-genbank-metadata -m NCBI-METADATA.txt \
-o NCBI-GENOMES \
--output-fasta-txt fasta.txt
Running this script will create the fasta.txt file, and populate the contents of the output directory NCBI-GENOMES with the following files for each genome:
- A FASTA file of contigs
- An external gene calls file (so that the PGAP gene calls can be imported into an anvi’o contigs database).
- An external functions file (so that the functions assigned to these genes can be imported into an anvi’o contigs database).
Some genomes may be missing external gene calls and functions depending on their state on the NCBI. Take a look at your fasta.txt and make sure each genome is associated with all three files.
If you are here, please familiarize yourself with the fasta.txt and the output directory NCBI-GENOMES by taking a quick look at them.
We can now use fasta.txt in a config file for anvi’o workflows for contigs, phylogenomics or pangenomics for high-throughput / convenient analyses.
Excluding the gene calls from the fasta.txt file
When you specify an external gene calls file in your fasta.txt file, then anvi-run-workflow will use these gene calls instead of running Prodigal.
In this case the “source” for the gene calls will be noted as “PGAP”. This means that we might end up having some of the genomes with gene calls from PGAP and others with gene calls from Prodigal.
But because of the problem descirbed in this github issue, when we run a pangenomic or phylogenomic analysis with anvi’o, all the genomes must contain gene calls from the same source.
In order to bypass this issue, this is how you can run anvi-script-process-genbank-metadata:
anvi-script-process-genbank-metadata -m NCBI-METADATA.txt \
-o NCBI-GENOMES \
--output-fasta-txt fasta.txt \
--exclude-gene-calls-from-fasta-txt
Alternatively, you can go rebel and edit the external gene calls files to replace the source PGAP with prodigal. But you must realize that in some cases this may yield misleading insights. But we can’t stop you from doing that and cheating computers can occasionally be OK for those who know what they’re doing.
The output directory would stay the same, but the fasta.txt now looks like this:
This is a good moment to fix those names to more readable unique names. For instance the first one could be “Ca_A_pacificus_JGI68” and would have looked much better in any interface.
$ column -t fasta.txt | head
name path gene_functional_annotation
Candidatus_Altimarinus_pacificus_JGI_0000068_E11_GCA_000405005.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Altimarinus_pacificus_JGI_0000068_E11_GCA_000405005.1-contigs.fa
Candidatus_Gracilibacteria_bacterium_CG12_big_fil_rev_8_21_14_0_65_38_15_GCA_002771035.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG12_big_fil_rev_8_21_14_0_65_38_15_GCA_002771035.1-contigs.fa /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG12_big_fil_rev_8_21_14_0_65_38_15_GCA_002771035.1-external-functions.txt
Candidatus_Gracilibacteria_bacterium_CG17_big_fil_post_rev_8_21_14_2_50_48_13_GCA_002783265.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG17_big_fil_post_rev_8_21_14_2_50_48_13_GCA_002783265.1-contigs.fa /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG17_big_fil_post_rev_8_21_14_2_50_48_13_GCA_002783265.1-external-functions.txt
Candidatus_Gracilibacteria_bacterium_CG18_big_fil_WC_8_21_14_2_50_38_16_GCA_002786835.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG18_big_fil_WC_8_21_14_2_50_38_16_GCA_002786835.1-contigs.fa /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG18_big_fil_WC_8_21_14_2_50_38_16_GCA_002786835.1-external-functions.txt
Candidatus_Gracilibacteria_bacterium_CG1_02_38_174_GCA_001871945.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG1_02_38_174_GCA_001871945.1-contigs.fa /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG1_02_38_174_GCA_001871945.1-external-functions.txt
Candidatus_Gracilibacteria_bacterium_CG2_30_37_12_GCA_001873165.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG2_30_37_12_GCA_001873165.1-contigs.fa /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG2_30_37_12_GCA_001873165.1-external-functions.txt
Candidatus_Gracilibacteria_bacterium_CG_4_10_14_0_8_um_filter_38_28_GCA_002785345.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG_4_10_14_0_8_um_filter_38_28_GCA_002785345.1-contigs.fa /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG_4_10_14_0_8_um_filter_38_28_GCA_002785345.1-external-functions.txt
Candidatus_Gracilibacteria_bacterium_CG_4_9_14_0_2_um_filter_38_7_GCA_002788335.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG_4_9_14_0_2_um_filter_38_7_GCA_002788335.1-contigs.fa /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_CG_4_9_14_0_2_um_filter_38_7_GCA_002788335.1-external-functions.txt
Candidatus_Gracilibacteria_bacterium_GCA_002746815.1 /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_GCA_002746815.1-contigs.fa /Users/alonshaiber/Downloads/NCBI-GENOMES/Candidatus_Gracilibacteria_bacterium_GCA_002746815.1-external-functions.txt
By excluding the external gene calls file from the fasta.txt file, we guarantee that anvi-run-workflow will use Prodigal for gene calls and there will be no disagreement on how genes were called between genomes from NCBI and genomes from other sources that are already in anvi’o.
If you have other FASTA files to include in any downstream analyses, you can include add them into this fasta.txt file.
Running snakemake workflows
If you would like to use the anvi’o snakemake workflows effectively, please first go through this tutorial.
We generally first run the contigs workflow on these files. You can get a default config file for the contigs workflow as described in the anvi’o snakemake workflow tutorial and take a look at all the parameters available to you. But the easiest thing to use would be this config file CONTIGS-CONFIG.json:
{
"fasta_txt": "fasta.txt"
}
Now we can run the following, which would create all the contigs databases (in 6 threads):
anvi-run-workflow -w contigs \
-c CONTIGS-CONFIG.json \
--additional-params \
--jobs 6 \
--resources nodes=6
Once this is done, one can go to any direction with the resulting contigs databases.
Instead, you can run the pangenomics workflow directly on these genomes with the following configuration file, and it would first automatically run the contigs workflow for you:
{
"fasta_txt": "fasta.txt",
"anvi_gen_contigs_database": {
"--project-name": "{group}",
"threads": 4
},
"anvi_run_hmms": {
"run": true,
"threads": 5
},
"anvi_run_kegg_kofams": {
"run": false
},
"anvi_run_ncbi_cogs": {
"run": true
},
"anvi_run_scg_taxonomy": {
"run": true
},
"anvi_run_trna_scan": {
"run": false
},
"anvi_script_reformat_fasta": {
"run": true,
"--min-len": "1000"
},
"anvi_pan_genome": {
"threads": 5
},
"project_name": "YOUR_PROJECT_NAME_HERE",
"internal_genomes": "",
"external_genomes": "external-genomes.txt",
"max_threads": "",
"config_version": "2",
"workflow_name": "pangenomics"
}
If you saved it as PAN-CONFIG.json, you can run the entire workflow like this:
anvi-run-workflow -w pangenomics \
-c PAN-CONFIG.json \
--additional-params \
--jobs 6 \
--resources nodes=6
Once this is done, you can display your pangenome using anvi-display-pan.